bactopia
Tags: bacteria assembly annotation amr mlst genomics pipeline named-workflow
Pipeline abrangente de análise bacteriana para caracterização genômica completa.
Este fluxo de trabalho realiza análises de ponta a ponta, incluindo controle de qualidade, montagem, anotação, detecção de resistência antimicrobiana, tipagem MLST e análise opcional específica de patógenos via Merlin. Ele processa reads de sequenciamento brutos e produz uma caracterização genômica completa adequada para análises posteriores.
Visão Geral do Pipeline
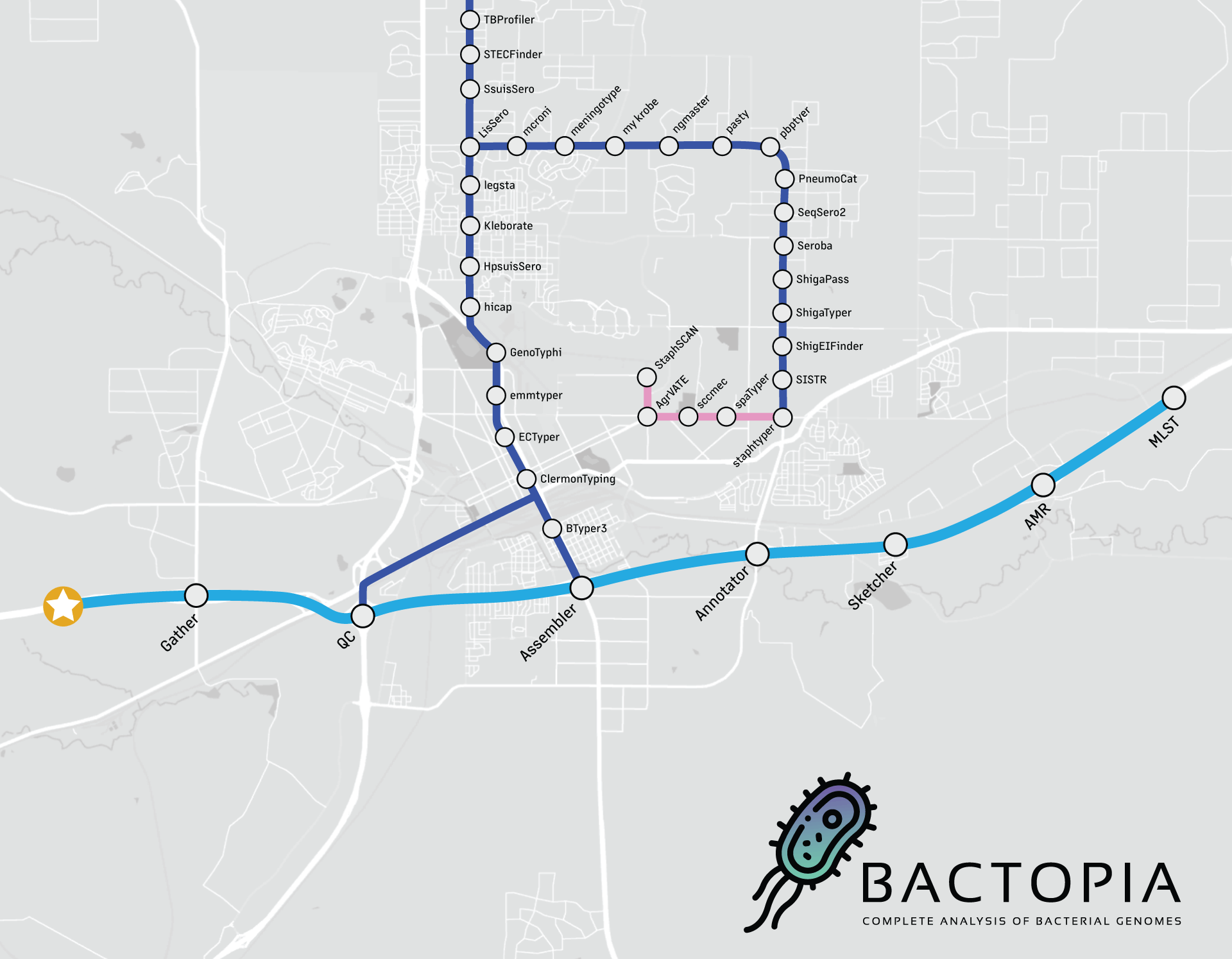
Ao olhar para o fluxo de trabalho acima, pode parecer que não há muita coisa acontecendo, mas posso garantir que muita coisa está ocorrendo. O fluxo de trabalho é dividido em 8 etapas:
- Gather - Coletar todos os dados em um único lugar
- QC - Controle de qualidade dos dados
- Assembler - Montar os reads em contigs
- Annotator - Anotar os contigs
- Sketcher - Criar um esboço dos contigs e consultar bancos de dados
- Sequence Typing - Determinar o tipo de sequência dos contigs
- Antibiotic Resistance - Determinar a resistência a antibióticos dos contigs e proteínas
- Merlin - Executar automaticamente ferramentas específicas por espécie com base na distância
Se você está procurando um guia para começar rapidamente, consulte o Guia para Iniciantes.
Etapa 1 - Gather
O objetivo principal da etapa gather é reunir todas as amostras em um único lugar. Isso
inclui o download de amostras do ENA/SRA ou NCBI Assembly. As ferramentas utilizadas são:
| Ferramenta | Descrição |
|---|---|
| art | Para simular reads sem erros a partir de uma montagem de entrada |
| fastq-dl | Download de arquivos FASTQ do ENA/SRA |
| ncbi-genome-download | Download de arquivos FASTA do NCBI Assembly |
A etapa gather também realiza verificações básicas de controle de qualidade para ajudar a prevenir falhas posteriores.
Verificações de Qualidade com Falha
| Nome do Arquivo | Descrição |
|---|---|
| -gzip-error.txt | Amostra falhou nas verificações de Gzip e foi excluída de análises posteriores |
| -low-basepair-proportion-error.txt | Amostra falhou nas verificações de proporção de pares de bases e foi excluída de análises posteriores |
| -low-read-count-error.txt | Amostra falhou nas verificações de contagem de reads e foi excluída de análises posteriores |
| -low-sequence-depth-error.txt | Amostra falhou nas verificações de pares de bases sequenciados e foi excluída de análises posteriores |
Amostras que falham em qualquer uma das verificações de qualidade serão excluídas de análises posteriores.
Essas amostras gerarão um arquivo *-error.txt com a mensagem de erro. Excluir
essas amostras previne falhas posteriores que causariam a falha de todo o fluxo de trabalho.
Exemplo de Erro: FASTQ(s) de entrada falharam nas verificações de Gzip
Se os FASTQ(s) de entrada não passarem no teste de Gzip, a amostra será excluída de análises posteriores.
Exemplo de texto em <SAMPLE_NAME>-gzip-error.txt <SAMPLE_NAME> FASTQs failed Gzip tests. Please check the input FASTQs. Further analysis is discontinued.
Exemplo de Erro: FASTQs de entrada têm número desproporcional de reads
Se os FASTQ(s) de entrada de uma amostra tiverem um número desproporcionalmente diferente de reads
entre os dois pares, a amostra será excluída de análises posteriores. Você pode
ajustar essa contagem mínima de reads usando o parâmetro --min_proportion.
Exemplo de texto em <SAMPLE_NAME>-low-basepair-proportion-error.txt
<SAMPLE_NAME> FASTQs failed to meet the minimum shared basepairs. They
shared Y basepairs, with R1 having A bp and R2 having B bp. Further
analysis is discontinued.
Exemplo de Erro: FASTQ(s) de entrada têm reads insuficientes
Se os FASTQ(s) de entrada de uma amostra tiverem menos reads do que o mínimo exigido, a
amostra será excluída de análises posteriores. Você pode ajustar essa contagem mínima de
reads usando o parâmetro --min_reads.
Exemplo de texto em <SAMPLE_NAME>-low-read-count-error.txt
<SAMPLE_NAME> FASTQ(s) contain X total reads. This does not exceed the required
minimum Y read count. Further analysis is discontinued.
Exemplo de Erro: FASTQ(s) de entrada têm pares de bases sequenciados insuficientes
Se os FASTQ(s) de entrada de uma amostra não atingirem o número mínimo de pares de bases
sequenciados, a amostra será excluída de análises posteriores. Você pode
ajustar essa contagem mínima usando o parâmetro --min_basepairs.
Exemplo de texto em <SAMPLE_NAME>-low-sequence-depth-error.txt
<SAMPLE_NAME> FASTQ(s) contain X total basepairs. This does not exceed the
required minimum Y bp. Further analysis is discontinued.
Etapa 2 - QC
O módulo qc usa uma variedade de ferramentas para realizar controle de qualidade em reads do Illumina e
Oxford Nanopore. As ferramentas utilizadas são:
| Ferramenta | Tecnologia | Descrição |
|---|---|---|
| bbtools | Illumina | Um conjunto de ferramentas para manipulação de reads |
| fastp | Illumina | Uma ferramenta projetada para fornecer pré-processamento rápido e completo de arquivos FastQ |
| fastqc | Illumina | Uma ferramenta de controle de qualidade para dados de sequenciamento de alto rendimento |
| fastq_scan | Nanopore | Uma ferramenta para varredura rápida de arquivos FASTQ |
| lighter | Illumina | Uma ferramenta para correção de erros de sequenciamento em reads do Illumina |
| NanoPlot | Nanopore | Uma ferramenta para visualização de dados de sequenciamento de leituras longas |
| nanoq | Nanopore | Uma ferramenta para calcular métricas de qualidade para reads do Oxford Nanopore |
| porechop | Nanopore | Uma ferramenta para remoção de adaptadores de reads do Oxford Nanopore |
| rasusa | Nanopore | Subamostragem aleatória de reads de sequenciamento para uma cobertura especificada |
Semelhante à etapa gather, a etapa qc também impedirá que amostras que não atendam às
verificações básicas de qualidade continuem para as etapas posteriores.
Verificações de Qualidade com Falha
| Nome do Arquivo | Descrição |
|---|---|
| .error-fastq.gz | Um arquivo FASTQ comprimido com reads que falharam no controle de qualidade |
| -low-read-count-error.txt | Amostra falhou nas verificações de contagem de reads e foi excluída de análises posteriores |
| -low-sequence-coverage-error.txt | Amostra falhou nas verificações de cobertura de sequenciamento e foi excluída de análises posteriores |
| -low-sequence-depth-error.txt | Amostra falhou nas verificações de pares de bases sequenciados e foi excluída de análises posteriores |
Amostras que falham em qualquer uma das verificações de qualidade serão excluídas de análises posteriores.
Essas amostras gerarão um arquivo *-error.txt com a mensagem de erro. Excluir
essas amostras previne falhas posteriores que causariam a falha de todo o fluxo de trabalho.
Exemplo de Erro: Após o controle de qualidade, restam reads insuficientes
Se após a limpeza dos reads, uma amostra tiver menos reads do que o mínimo exigido, a
amostra será excluída de análises posteriores. Você pode ajustar essa contagem mínima de
reads usando o parâmetro --min_reads.
Exemplo de texto em <SAMPLE_NAME>-low-read-count-error.txt
<SAMPLE_NAME> FASTQ(s) contain X total reads. This does not exceed the required
minimum Y read count. Further analysis is discontinued.
Exemplo de Erro: Após o controle de qualidade, resta cobertura de sequenciamento insuficiente
Se após a limpeza dos reads, uma amostra não atingir a cobertura mínima de sequenciamento
exigida, a amostra será excluída de análises posteriores. Você pode
ajustar esse mínimo usando o parâmetro --min_coverage.
Nota: Esta verificação é realizada apenas quando o tamanho do genoma está disponível.
Exemplo de texto em <SAMPLE_NAME>-low-sequence-coverage-error.txt
After QC, <SAMPLE_NAME> FASTQ(s) contain X total basepairs. This does not
exceed the required minimum Y bp (Zx coverage). Further analysis is
discontinued.
Exemplo de Erro: Após o controle de qualidade, restam pares de bases sequenciados insuficientes
Se após a limpeza dos reads, uma amostra não atingir o número mínimo de pares de bases
sequenciados, a amostra será excluída de análises posteriores. Você pode
ajustar esse mínimo usando o parâmetro --min_basepairs.
Exemplo de texto em <SAMPLE_NAME>-low-sequence-depth-error.txt
<SAMPLE_NAME> FASTQ(s) contain X total basepairs. This does not exceed the
required minimum Y bp. Further analysis is discontinued.
Etapa 3 - Assembler
O módulo assembler usa uma variedade de ferramentas de montagem para criar uma montagem de
reads do Illumina e Oxford Nanopore. As ferramentas utilizadas são:
| Ferramenta | Descrição |
|---|---|
| Dragonflye | Montagem de reads do Oxford Nanopore, bem como montagem híbrida com polimento por reads curtos |
| Shovill | Montagem de reads paired-end do Illumina |
| Shovill-SE | Montagem de reads single-end do Illumina |
| Unicycler | Montagem híbrida, utilizando primeiro reads curtos e depois reads longos |
Estatísticas resumidas para cada montagem são geradas usando assembly-scan.
--short_polish em vez de --hybrid com sequenciamento ONT recenteUsar Unicycler (--hybrid) para criar uma montagem híbrida
funciona muito bem quando você tem reads longos com baixa cobertura e muito ruído. No entanto, se você estiver
usando sequenciamento ONT recente, provavelmente tem alta cobertura e o uso do método --short_polish
vai gerar resultados melhores (e mais rápidos!) do que --hybrid.
Verificações de Qualidade com Falha
| Nome do Arquivo | Descrição |
|---|---|
| -assembly-error.txt | Amostra falhou nas verificações de montagem e foi excluída de análises posteriores |
Amostras que falham em qualquer uma das verificações de qualidade serão excluídas de análises posteriores.
Essas amostras gerarão um arquivo *-error.txt com a mensagem de erro. Excluir
essas amostras previne falhas posteriores que causariam a falha de todo o fluxo de trabalho.
Exemplo de Erro: Montagem realizada com sucesso, mas 0 contigs
Se uma amostra for montada com sucesso, mas 0 contigs forem formados, a amostra será excluída de análises posteriores.
Exemplo de texto em <SAMPLE_NAME>-assembly-error.txt <SAMPLE_NAME> assembled successfully, but 0 contigs were formed. Please investigate <SAMPLE_NAME> to determine a cause (e.g. metagenomic, contaminants, etc...) for this outcome. Further assembly-based analysis of <SAMPLE_NAME> will be discontinued.
Exemplo de Erro: Montagem realizada com sucesso, mas tamanho da montagem ruim
Se sua amostra for montada com sucesso, mas o tamanho da montagem for menor do que o tamanho
mínimo permitido do genoma, a amostra será excluída de análises posteriores. Você pode
ajustar esse tamanho mínimo usando o parâmetro --min_genome_size.
Exemplo de texto em <SAMPLE_NAME>-assembly-error.txt
<SAMPLE_NAME> assembled size (000 bp) is less than the minimum allowed genome
size (000 bp). If this is unexpected, please investigate <SAMPLE_NAME> to
determine a cause (e.g. metagenomic, contaminants, etc...) for the poor assembly.
Otherwise, adjust the --min_genome_size parameter to fit your need. Further
assembly based analysis of <SAMPLE_NAME> will be discontinued.
Etapa 4 - Annotator
A etapa annotator usa Prokka (padrão)
ou Bakta (via --use_bakta) para anotar
contigs montados com informações funcionais incluindo genes, proteínas, rRNA, tRNA
e outras características genômicas.
Etapa 5 - Sketcher
O módulo sketcher usa Mash e
Sourmash para criar esboços e consultar
RefSeq e GTDB.
Etapa 6 - Sequence Typing
A etapa mlst usa mlst para varrer montagens contra
esquemas de tipagem do PubMLST e determinar o tipo de sequência.
Etapa 7 - Antibiotic Resistance
A etapa amrfinderplus usa AMRFinder+ para identificar
genes de resistência antimicrobiana e mutações pontuais tanto de contigs montados quanto de
sequências de proteínas anotadas.
Etapa 8 - Merlin
A etapa merlin seleciona e executa automaticamente ferramentas de tipagem específicas por espécie com base
nos resultados de distância Mash da etapa sketcher. Ative com --ask_merlin. Consulte a
seção de saídas de Análise Específica de Patógenos abaixo para a
lista completa de organismos e ferramentas suportados.
Uso
Bactopia CLI:
bactopia \
--input samples.csv \
--outdir results/
Nextflow:
nextflow run bactopia/bactopia \
--input samples.csv \
--outdir results/
Saídas
Arquivos de Saída Esperados
<BACTOPIA_DIR>
├── <SAMPLE_NAME>
│ ├── main
│ │ ├── annotator
│ │ │ └── prokka
│ │ │ ├── <SAMPLE_NAME>-blastdb.tar.gz
│ │ │ ├── <SAMPLE_NAME>.faa.gz
│ │ │ ├── <SAMPLE_NAME>.ffn.gz
│ │ │ ├── <SAMPLE_NAME>.fna.gz
│ │ │ ├── <SAMPLE_NAME>.fsa.gz
│ │ │ ├── <SAMPLE_NAME>.gbk.gz
│ │ │ ├── <SAMPLE_NAME>.gff.gz
│ │ │ ├── <SAMPLE_NAME>.sqn.gz
│ │ │ ├── <SAMPLE_NAME>.tbl.gz
│ │ │ ├── <SAMPLE_NAME>.tsv
│ │ │ ├── <SAMPLE_NAME>.txt
│ │ │ └── logs
│ │ │ ├── <SAMPLE_NAME>.err
│ │ │ ├── <SAMPLE_NAME>.log
│ │ │ ├── nf.command.{begin,err,log,out,run,sh,trace}
│ │ │ └── versions.yml
│ │ ├── assembler
│ │ │ ├── <SAMPLE_NAME>.fna.gz
│ │ │ ├── <SAMPLE_NAME>.tsv
│ │ │ ├── logs
│ │ │ │ ├── nf.command.{begin,err,log,out,run,sh,trace}
│ │ │ │ ├── shovill-se.log
│ │ │ │ └── versions.yml
│ │ │ └── supplemental
│ │ │ ├── illumina.txt
│ │ │ └── shovill.corrections
│ │ ├── gather
│ │ │ ├── <SAMPLE_NAME>-meta.tsv
│ │ │ └── logs
│ │ │ ├── nf.command.{begin,err,log,out,run,sh,trace}
│ │ │ └── versions.yml
│ │ ├── qc
│ │ │ ├── <SAMPLE_NAME>_SE.fastq.gz
│ │ │ ├── logs
│ │ │ │ ├── <SAMPLE_NAME>-fastp.log
│ │ │ │ ├── nf.command.{begin,err,log,out,run,sh,trace}
│ │ │ │ └── versions.yml
│ │ │ └── supplemental
│ │ │ ├── <SAMPLE_NAME>.fastp.html
│ │ │ ├── <SAMPLE_NAME>.fastp.json
│ │ │ ├── <SAMPLE_NAME>_SE-final.json
│ │ │ ├── <SAMPLE_NAME>_SE-final_fastqc.html
│ │ │ ├── <SAMPLE_NAME>_SE-final_fastqc.zip
│ │ │ ├── <SAMPLE_NAME>_SE-original.json
│ │ │ ├── <SAMPLE_NAME>_SE-original_fastqc.html
│ │ │ └── <SAMPLE_NAME>_SE-original_fastqc.zip
│ │ └── sketcher
│ │ ├── <SAMPLE_NAME>-k21.msh
│ │ ├── <SAMPLE_NAME>-k31.msh
│ │ ├── <SAMPLE_NAME>-mash-refseq88-k21.txt
│ │ ├── <SAMPLE_NAME>-sourmash-gtdb-rs207-k31.txt
│ │ ├── <SAMPLE_NAME>.sig
│ │ └── logs
│ │ ├── nf.command.{begin,err,log,out,run,sh,trace}
│ │ └── versions.yml
│ └── tools
│ ├── amrfinderplus
│ │ ├── <SAMPLE_NAME>.tsv
│ │ └── logs
│ │ ├── nf.command.{begin,err,log,out,run,sh,trace}
│ │ └── versions.yml
│ └── mlst
│ ├── <SAMPLE_NAME>.tsv
│ └── logs
│ ├── nf.command.{begin,err,log,out,run,sh,trace}
│ └── versions.yml
└── bactopia-runs
└── bactopia-<TIMESTAMP>
├── merged-results
│ ├── amrfinderplus.tsv
│ ├── assembly-scan.tsv
│ ├── logs
│ │ ├── amrfinderplus-concat
│ │ │ ├── nf.command.{begin,err,log,out,run,sh,trace}
│ │ │ └── versions.yml
│ │ ├── assembly-scan-concat
│ │ │ ├── nf.command.{begin,err,log,out,run,sh,trace}
│ │ │ └── versions.yml
│ │ ├── meta-concat
│ │ │ ├── nf.command.{begin,err,log,out,run,sh,trace}
│ │ │ └── versions.yml
│ │ └── mlst-concat
│ │ ├── nf.command.{begin,err,log,out,run,sh,trace}
│ │ └── versions.yml
│ ├── meta.tsv
│ └── mlst.tsv
└── nf-reports
├── bactopia-dag.dot
├── bactopia-report.html
└── bactopia-timeline.html
Controle de Qualidade
| Arquivo | Descrição |
|---|---|
supplemental/*_fastqc.* | Relatórios de controle de qualidade do FastQC para reads brutos e limpos |
supplemental/*-NanoPlot.* | Relatórios do NanoPlot para reads do Nanopore |
supplemental/*.fastp.* | Relatórios de qualidade do Fastp (quando aplicável) |
supplemental/*_original.json | Métricas de qualidade para reads originais |
supplemental/*_final.json | Métricas de qualidade para reads finais |
Montagem
| Arquivo | Descrição |
|---|---|
*.fasta | Sequências do genoma montado |
assembly-stats.tsv | Métricas de qualidade da montagem |
merged-assembly-stats.tsv | Estatísticas consolidadas de montagem |
Anotação
O formato de saída depende da ferramenta de anotação escolhida (Bakta ou Prokka)
| Arquivo | Descrição |
|---|---|
*.gff.gz | Anotação do genoma no formato GFF3 (comprimida) |
*.gbk.gz | Anotação do genoma no formato GenBank (comprimida) |
*.faa.gz | Sequências de proteínas (comprimidas) |
*.fna.gz | Sequências de nucleotídeos da anotação (comprimidas) |
*.ffn.gz | Sequências de nucleotídeos de características (comprimidas) |
annotation.tsv | Tabelas de resumo de anotação |
blastdb.* | Banco de dados BLAST criado a partir da anotação |
Tipagem
| Arquivo | Descrição |
|---|---|
mlst.tsv | Resultados do tipo de sequência MLST |
merged-mlst.tsv | Resultados consolidados de MLST |
Resistência Antimicrobiana
| Arquivo | Descrição |
|---|---|
amrfinderplus.tsv | Resultados de detecção de genes de resistência antimicrobiana |
amrfinderplus.mutation.tsv | Resultados de mutações pontuais de resistência antimicrobiana |
merged-amrfinderplus.tsv | Resultados consolidados de resistência antimicrobiana |
Análise Comparativa
| Arquivo | Descrição |
|---|---|
*-k21.msh | Arquivos de esboço Mash (k=21) |
*-k31.msh | Arquivos de esboço Mash (k=31) |
*-mash-refseq88-*.txt | Resultados de triagem Mash contra RefSeq |
*.sig | Assinaturas do Sourmash |
sourmash-*.txt | Resultados de classificação do Sourmash |
Análise Específica de Patógenos
Criada apenas se --ask_merlin estiver ativado
| Arquivo | Descrição |
|---|---|
merlin/clermontyping/* | Tipagem de filogrupo de E. coli |
merlin/ectyper/* | Tipagem de E. coli enterotoxigênica |
merlin/shigatyper/* | Predição de sorotipo de Shigella |
merlin/shigapass/* | Vigilância passiva de Shigella |
merlin/shigeifinder/* | Detecção de Shigella e EIEC |
merlin/stecfinder/* | Detecção e tipagem de STEC |
merlin/emmtyper/* | Tipagem emm de S. pyogenes |
merlin/hicap/* | Tipagem capsular de H. influenzae |
merlin/hpsuissero/* | Sorotipagem de H. parasuis |
merlin/kleborate/* | Tipagem de espécies de Klebsiella |
merlin/staphtyper/* | Tipagem spa de S. aureus |
merlin/agrvate/* | Tipagem agr de S. aureus |
merlin/sccmec/* | Tipagem SCCmec de S. aureus |
Resultados Consolidados
Resultados agregados no nível de execução de todas as amostras
| Arquivo | Descrição |
|---|---|
samplesheet.tsv | Metadados das amostras e métricas de qualidade |
Trilha de Auditoria
Abaixo estão os arquivos que podem ajudá-lo a entender quais parâmetros e versões de programas foram utilizados.
Logs
Cada processo executado terá uma pasta chamada logs. Nessa pasta há arquivos úteis
para você revisar caso necessário.
| Extensão | Descrição |
|---|---|
| .begin | Um arquivo vazio usado para indicar que o processo foi iniciado |
| .err | Contém saídas STDERR do processo |
| .log | Contém saídas STDERR e STDOUT do processo |
| .out | Contém saídas STDOUT do processo |
| .run | O script que o Nextflow usa para preparar/desfazer o staging de arquivos e enfileirar processos com base no perfil definido |
| .sh | O script executado pelo bash para o processo |
| .trace | O relatório de rastreamento do Nextflow para o processo |
| versions.yml | Um arquivo no formato YAML com as versões dos programas |
Relatórios do Nextflow
Esses relatórios do Nextflow fornecem um excelente resumo da sua execução. Eles podem ser usados para otimizar o uso de recursos e estimar custos esperados ao utilizar plataformas em nuvem.
| Nome do Arquivo | Descrição |
|---|---|
| bactopia-dag.dot | A visualização DAG do Nextflow |
| bactopia-report.html | O Relatório de Execução do Nextflow |
| bactopia-timeline.html | O Relatório de Linha do Tempo do Nextflow |
| bactopia-trace.txt | O relatório de Rastreamento do Nextflow |
Parâmetros
Parâmetros Obrigatórios
Os parâmetros a seguir são a forma como você fornecerá amostras locais ou remotas para serem processadas pelo Bactopia.
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--samples | string | Um FOFN (via bactopia prepare) com nomes de amostras e caminhos para FASTQ/FASTAs a serem processados | |
--r1 | string | Primeiro conjunto de reads paired-end do Illumina comprimidos (gzip) (requer --r2 e --sample) | |
--r2 | string | Segundo conjunto de reads paired-end do Illumina comprimidos (gzip) (requer --r1 e --sample) | |
--se | string | Reads single-end do Illumina comprimidos (gzip) (requer --sample) | |
--ont | string | Reads do Oxford Nanopore comprimidos (gzip) (requer --sample) | |
--hybrid | boolean | false | Criar montagem híbrida usando Unicycler (requer --r1, --r2, --ont e --sample) |
--short_polish | boolean | false | Criar montagem híbrida a partir de montagem de reads longos com polimento por reads curtos (requer --r1, --r2, --ont e --sample) |
--sample | string | Nome da amostra a ser usado para as sequências de entrada | |
--accessions | string | Um arquivo contendo accessions de Experimentos ENA/SRA ou accessions de NCBI Assembly a serem processados | |
--accession | string | Nome da amostra a ser usado para as sequências de entrada | |
--assembly | string | Um genoma montado em formato FASTA comprimido (requer --sample) | |
--check_samples | boolean | false | Validar o FOFN de entrada fornecido por --samples |
Parâmetros do AMRFinder+
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--amrfinderplus_ident_min | number | -1 | Proporção mínima de aminoácidos idênticos no alinhamento para um hit (0..1) |
--amrfinderplus_coverage_min | number | 0.5 | Cobertura mínima da proteína de referência (0..1) |
--amrfinderplus_organism | string | Grupo taxonômico para executar triagens adicionais | |
--amrfinderplus_translation_table | integer | 11 | Código genético NCBI para BLAST traduzido |
--amrfinderplus_noplus | boolean | false | Desativar a execução do AMRFinder+ com a opção --plus |
--amrfinderplus_report_common | boolean | false | Reportar proteínas comuns a um grupo taxonômico |
--amrfinderplus_report_all_equal | boolean | false | Reportar todos os hits BLAST e HMM com pontuação igual |
--amrfinderplus_opts | string | Opções extras do AMRFinder+ entre aspas | |
--amrfinderplus_db | string | Um banco de dados AMRFinder+ personalizado a ser usado, seja um tarball ou uma pasta |
Parâmetros do csvtk concat
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--csvtk_concat_opts | string | Opções extras do csvtk concat entre aspas |
Parâmetros do Assembler
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--shovill_assembler | string | skesa | Montador a ser usado pelo Shovill (opções: skesa, megahit, spades, velvet) |
--dragonflye_assembler | string | flye | Montador a ser usado pelo Dragonflye (opções: flye, miniasm, raven) |
--use_unicycler | boolean | Usar Unicycler para montagem paired-end | |
--min_contig_len | integer | 500 | Comprimento mínimo de contig <0=AUTO> |
--min_contig_cov | integer | 2 | Cobertura mínima de contig <0=AUTO> |
--contig_namefmt | string | Formato dos IDs FASTA dos contigs no estilo 'printf' | |
--shovill_opts | string | Opções extras do montador entre aspas para o Shovill | |
--shovill_kmers | string | K-mers a serem usados <em branco=AUTO> | |
--dragonflye_opts | string | Opções extras do montador entre aspas para o Dragonflye | |
--trim | boolean | Ativar trimagem de adaptadores | |
--no_stitch | boolean | Desativar costura de reads para reads paired-end | |
--no_corr | boolean | Desativar correção pós-montagem | |
--unicycler_mode | string | normal | Modo de bridging usado pelo Unicycler (opções: conservative, normal, bold) |
--min_component_size | integer | 1000 | Extremidades mortas do grafo menores que este tamanho (bp) serão removidas do grafo final |
--min_dead_end_size | integer | 1000 | Extremidades mortas do grafo menores que este tamanho (bp) serão removidas do grafo final |
--nanohq | boolean | false | Para o Flye, usar '--nano-hq' em vez de --nano-raw |
--medaka_model | string | O modelo a ser usado para polimento com Medaka | |
--medaka_rounds | integer | 0 | O número de rodadas de polimento com Medaka a realizar |
--racon_rounds | integer | 1 | O número de rodadas de polimento com Racon a realizar |
--no_polish | boolean | Pular a etapa de polimento da montagem | |
--no_miniasm | boolean | Pular bridging com miniasm+Racon | |
--no_rotate | boolean | Não rotacionar replicons completos para iniciar em um gene padrão | |
--reassemble | boolean | false | Se os reads foram simulados, eles serão usados para criar uma nova montagem |
--polypolish_rounds | integer | 1 | Número de rodadas de polimento a realizar com Polypolish para polimento por reads curtos |
--pilon_rounds | integer | 0 | Número de rodadas de polimento a realizar com Pilon para polimento por reads curtos |
Parâmetros do Gather
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--skip_fastq_check | boolean | Pular verificações de requisitos mínimos para FASTQs de entrada | |
--min_basepairs | integer | 2241820 | A quantidade mínima de pares de bases necessária para continuar as análises posteriores |
--min_reads | integer | 7472 | A quantidade mínima de reads necessária para continuar as análises posteriores |
--min_coverage | integer | 10 | A cobertura mínima necessária para continuar as análises posteriores |
--min_proportion | number | 0.5 | A proporção mínima de pares de bases para reads paired-end continuarem as análises posteriores |
--min_genome_size | integer | 100000 | O tamanho mínimo estimado do genoma permitido para a sequência de entrada continuar as análises posteriores |
--max_genome_size | integer | 18040666 | O tamanho máximo estimado do genoma permitido para a sequência de entrada continuar as análises posteriores |
--attempts | integer | 3 | Número máximo de tentativas de download |
--use_ena | boolean | Baixar FASTQs do ENA | |
--no_cache | boolean | Pular o cache do arquivo de resumo de montagem do ncbi-genome-download |
Parâmetros do Sketcher
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--sketch_size | integer | 10000 | Tamanho do esboço. Cada esboço terá no máximo este número de min-hashes não redundantes |
--sourmash_scale | integer | 10000 | Escolher o número de hashes como 1 em uma FRAÇÃO dos k-mers de entrada |
--no_winner_take_all | boolean | Desativar a estratégia winner-takes-all para estimativas de identidade | |
--screen_i | number | 0.8 | Identidade mínima para reportar |
Parâmetros do MLST
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--mlst_scheme | string | Não detectar automaticamente, forçar este esquema em todas as entradas | |
--mlst_minid | integer | 95 | Porcentagem mínima de identidade de DNA do alelo completo para considerar 'similar' |
--mlst_mincov | integer | 10 | Porcentagem mínima de cobertura de DNA para reportar alelo parcial |
--mlst_minscore | integer | 50 | Pontuação mínima de 100 para corresponder a um esquema |
--mlst_nopath | boolean | false | Remover caminhos de arquivo da coluna FILE |
--mlst_db | string | Um banco de dados MLST personalizado a ser usado, seja um tarball ou um diretório |
Parâmetros do QC
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--use_bbmap | boolean | Reads do Illumina serão processados com BBMap | |
--use_porechop | boolean | false | Usar Porechop para remover adaptadores de reads ONT |
--skip_qc | boolean | A etapa de controle de qualidade será ignorada e assumirá que as sequências de entrada já foram submetidas ao controle de qualidade | |
--skip_qc_plots | boolean | A criação de gráficos de controle de qualidade pelo FastQC ou Nanoplot será ignorada | |
--skip_error_correction | boolean | A correção de erros de reads pelo FLASH será ignorada | |
--adapters | string | Um arquivo FASTA contendo adaptadores a serem removidos | |
--adapter_k | integer | 23 | Comprimento do k-mer usado para encontrar adaptadores |
--phix | string | Genoma de referência do phiX174 para remoção | |
--phix_k | integer | 31 | Comprimento do k-mer usado para encontrar o phiX174 |
--ktrim | string | r | Trimar reads para remover bases correspondentes a k-mers de referência (opções: f, r, l) |
--mink | integer | 11 | Procurar k-mers mais curtos nas extremidades dos reads até este comprimento ao trimar ou mascarar por k-mer |
--hdist | integer | 1 | Distância máxima de Hamming para k-mers de referência (apenas substituições) |
--tpe | string | t | Ao trimar pelo lado direito do k-mer, trimar ambos os reads para o comprimento mínimo de qualquer um (opções: f, t) |
--tbo | string | t | Trimar adaptadores com base em onde os reads emparelhados se sobrepõem (opções: f, t) |
--qtrim | string | rl | Trimar extremidades dos reads para remover bases com qualidade abaixo de trimq (opções: rl, f, r, l, w) |
--trimq | integer | 6 | Regiões com qualidade média ABAIXO deste valor serão trimadas se qtrim estiver definido como algo diferente de f |
--maq | integer | 10 | Reads com qualidade média (após a trimagem) abaixo deste valor serão descartados |
--minlength | integer | 35 | Reads mais curtos que este valor após a trimagem serão descartados |
--ftm | integer | 5 | Se positivo, trimar o comprimento pelo lado direito para ser igual a zero, módulo deste número |
--tossjunk | string | t | Descartar reads com caracteres inválidos como bases (opções: f, t) |
--ain | string | f | Ao detectar nomes de pares, permitir nomes idênticos (opções: f, t) |
--qout | string | 33 | Offset PHRED a ser usado para FASTQs de saída (opções: 33, 64) |
--maxcor | integer | 1 | Número máximo de correções em uma janela de 20bp |
--sampleseed | integer | 42 | Definir um número positivo para usar como semente do gerador de números aleatórios para amostragem |
--ont_minlength | integer | 1000 | Reads ONT menores que este valor serão descartados |
--ont_minqual | integer | 0 | Filtro mínimo de qualidade média de reads ONT |
--porechop_opts | string | Opções extras do Porechop entre aspas | |
--nanoplot_opts | string | Opções extras do NanoPlot entre aspas | |
--bbduk_opts | string | Opções extras do BBDuk entre aspas | |
--fastp_opts | string | Opções extras do fastp entre aspas |
Parâmetros de Download do Bakta
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--bakta_db | string | Tarball ou caminho para o banco de dados do Bakta | |
--bakta_db_type | string | full | Qual BD do Bakta baixar: 'full' (~30GB) ou 'light' (~2GB) (opções: full, light) |
--bakta_save_as_tarball | boolean | false | Salvar o banco de dados do Bakta como tarball |
--download_bakta | boolean | false | Baixar o banco de dados do Bakta para o caminho especificado em --bakta_db |
Parâmetros do Bakta
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--bakta_proteins | string | Arquivo FASTA de proteínas confiáveis para anotar primeiro | |
--bakta_prodigal_tf | string | Arquivo de treinamento a ser usado para o Prodigal | |
--bakta_replicons | string | Tabela de informações de replicons (tsv/csv) | |
--bakta_min_contig_length | integer | 1 | Tamanho mínimo de contig para anotar |
--bakta_keep_contig_headers | boolean | false | Manter os cabeçalhos originais dos contigs |
--bakta_compliant | boolean | false | Forçar conformidade com Genbank/ENA/DDJB |
--bakta_skip_trna | boolean | false | Pular detecção e anotação de tRNA |
--bakta_skip_tmrna | boolean | false | Pular detecção e anotação de tmRNA |
--bakta_skip_rrna | boolean | false | Pular detecção e anotação de rRNA |
--bakta_skip_ncrna | boolean | false | Pular detecção e anotação de ncRNA |
--bakta_skip_ncrna_region | boolean | false | Pular detecção e anotação de regiões de ncRNA |
--bakta_skip_crispr | boolean | false | Pular detecção e anotação de arrays CRISPR |
--bakta_skip_cds | boolean | false | Pular detecção e anotação de CDS |
--bakta_skip_sorf | boolean | false | Pular detecção e anotação de sORF |
--bakta_skip_gap | boolean | false | Pular detecção e anotação de gaps |
--bakta_skip_ori | boolean | false | Pular detecção e anotação de oriC/oriT |
--bakta_opts | string | Opções extras do Bakta entre aspas. Exemplo: '--gram +' |
Parâmetros do Prokka
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--prokka_proteins | string | ${projectDir}/data/proteins.faa | Arquivo FASTA de proteínas confiáveis para anotar primeiro |
--prokka_prodigal_tf | string | Arquivo de treinamento a ser usado para o Prodigal | |
--prokka_compliant | boolean | false | Forçar conformidade com Genbank/ENA/DDJB |
--prokka_centre | string | Bactopia | ID do centro de sequenciamento |
--prokka_coverage | integer | 80 | Cobertura mínima na proteína de consulta |
--prokka_evalue | string | 1e-09 | Corte do e-value de similaridade |
--prokka_opts | string | Opções extras do Prokka entre aspas | |
--prokka_debug | boolean | false | Ativar modo de depuração para o Prokka |
Parâmetros do mashdist
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--mash_sketch | string | A sequência de referência como um Mash Sketch (arquivo .msh) | |
--mash_seed | integer | 42 | Semente a fornecer à função de hash |
--mash_table | boolean | false | Saída em formato de tabela (campos ficarão em branco se não atingirem o limiar do p-value) |
--mash_m | integer | 1 | Cópias mínimas de cada k-mer necessárias para passar o filtro de ruído para reads |
--mash_w | number | 0.01 | Limiar de probabilidade para avisar sobre tamanho de k-mer baixo |
--mash_max_p | number | 1.0 | P-value máximo a reportar |
--mash_max_dist | number | 1.0 | Distância máxima a reportar |
--merlin_dist | number | 0.1 | Distância máxima a reportar ao usar o Merlin |
--full_merlin | boolean | false | Executar o Merlin completo e todas as ferramentas específicas por espécie, independentemente da distância Mash |
--mash_use_fastqs | boolean | false | Consultar usando FASTQs em vez das montagens |
Parâmetros do ClermonTyping
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--clermontyping_threshold | integer | 0 | Não usar contigs abaixo deste tamanho |
Parâmetros do ECTyper
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--ectyper_opid | integer | 90 | Porcentagem de identidade exigida para uma correspondência de alelo do antígeno O |
--ectyper_opcov | integer | 90 | Porcentagem mínima de cobertura exigida para uma correspondência de alelo do antígeno O |
--ectyper_hpid | integer | 95 | Porcentagem de identidade exigida para uma correspondência de alelo do antígeno H |
--ectyper_hpcov | integer | 50 | Porcentagem mínima de cobertura exigida para uma correspondência de alelo do antígeno H |
--ectyper_verify | boolean | false | Ativar verificação de espécie de E. coli |
--ectyper_print_alleles | boolean | false | Imprime as sequências dos alelos como coluna final, se ativado |
Parâmetros do emmtyper
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--emmtyper_wf | string | blast | Fluxo de trabalho para o emmtyper usar (opções: blast, pcr) |
--emmtyper_blastdb | string | Caminho para um banco de dados EMM BLAST personalizado | |
--emmtyper_cluster_distance | integer | 500 | Distância entre cluster de correspondências para considerar como clusters diferentes |
--emmtyper_percid | integer | 95 | Porcentagem mínima de identidade da sequência |
--emmtyper_culling_limit | integer | 5 | Total de hits a retornar em uma posição |
--emmtyper_mismatch | integer | 5 | Limiar para número de incompatibilidades permitidas no hit BLAST |
--emmtyper_align_diff | integer | 5 | Limiar para diferença entre o comprimento do alinhamento e o comprimento do sujeito no BLAST |
--emmtyper_gap | integer | 2 | Limiar de gap permitido no hit BLAST |
--emmtyper_min_perfect | integer | 15 | Tamanho mínimo de correspondência perfeita na extremidade 3' do primer |
--emmtyper_min_good | integer | 15 | Tamanho mínimo onde deve haver 2 correspondências para cada incompatibilidade |
--emmtyper_max_size | integer | 2000 | Tamanho máximo do produto de PCR |
Parâmetros do hicap
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--hicap_database_dir | string | Diretório contendo o banco de dados de loci | |
--hicap_model_fp | string | Caminho para o modelo do Prodigal | |
--hicap_full_sequence | boolean | false | Escrever a sequência de entrada completa no arquivo genbank em vez de apenas a região ao redor e incluindo o locus |
--hicap_debug | boolean | false | hicap imprimirá mensagens de depuração |
--hicap_gene_coverage | number | 0.8 | Porcentagem mínima de cobertura para considerar um gene único completo |
--hicap_gene_identity | number | 0.7 | Porcentagem mínima de identidade para considerar um gene único completo |
--hicap_broken_gene_length | integer | 60 | Comprimento mínimo para considerar um gene quebrado |
--hicap_broken_gene_identity | number | 0.8 | Porcentagem mínima de identidade para considerar um gene quebrado |
Parâmetros do Mykrobe
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--mykrobe_species | string | Painel de espécies a usar (opções: sonnei, staph, tb, typhi) | |
--mykrobe_kmer | integer | 21 | Comprimento do k-mer |
--mykrobe_min_depth | integer | 1 | Profundidade mínima |
--mykrobe_model | string | kmer_count | Modelo de genótipo usado (opções: kmer_count, median_depth) |
--mykrobe_report_all_calls | boolean | false | Reportar todas as chamadas |
--mykrobe_opts | string | Opções extras do Mykrobe entre aspas |
Parâmetros do GenoTyphi
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--genotyphi_kmer | integer | 21 | Comprimento do k-mer |
--genotyphi_min_depth | integer | 1 | Profundidade mínima |
--genotyphi_model | string | kmer_count | Modelo de genótipo usado (opções: kmer_count, median_depth) |
--genotyphi_report_all_calls | boolean | false | Reportar todas as chamadas |
--genotyphi_mykrobe_opts | string | Opções extras do Mykrobe entre aspas |
Parâmetros do Kleborate
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--kleborate_preset | string | kpsc | Módulo predefinido a usar para o Kleborate (opções: kpsc, kosc, escherichia) |
--kleborate_opts | string | Opções extras entre aspas para o Kleborate |
Parâmetros do legsta
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--legsta_noheader | boolean | false | Não imprimir a linha de cabeçalho |
Parâmetros do LisSero
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--lissero_min_id | number | 95.0 | Porcentagem mínima de identidade para aceitar uma correspondência |
--lissero_min_cov | number | 95.0 | Cobertura mínima do gene para aceitar uma correspondência |
Parâmetros do ngmaster
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--ngmaster_csv | boolean | false | Gerar saída no formato separado por vírgula (CSV) em vez de separado por tabulação |
Parâmetros do pasty
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--pasty_min_pident | integer | 95 | Porcentagem mínima de identidade para contar um hit |
--pasty_min_coverage | integer | 95 | Porcentagem mínima de cobertura para contar um hit |
Parâmetros do pbptyper
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--pbptyper_min_pident | integer | 95 | Porcentagem mínima de identidade para contar um hit |
--pbptyper_min_coverage | integer | 95 | Porcentagem mínima de cobertura para contar um hit |
Parâmetros do SeqSero2
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--seqsero2_run_mode | string | k | Fluxo de trabalho a executar: modo 'a' de alelo ou modo 'k' de k-mer (opções: a, k) |
--seqsero2_input_type | string | assembly | Formato de entrada a analisar: 'assembly' ou 'fastq' (opções: assembly, fastq) |
--seqsero2_bwa_mode | string | mem | Algoritmos para mapeamento bwa no modo de alelo (opções: mem, sam) |
Parâmetros do SeroBA
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--seroba_noclean | boolean | false | Não limpar arquivos intermediários |
--seroba_coverage | integer | 20 | Limiar para cobertura de k-mer da sequência de referência |
Parâmetros do SISTR
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--sistr_full_cgmlst | boolean | false | Usar o conjunto completo de alelos cgMLST, que pode incluir alelos altamente similares |
Parâmetros do AgrVATE
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--agrvate_typing_only | boolean | false | Apenas tipagem agr. Pula a extração do operon agr e a detecção de frameshift |
Parâmetros do spaTyper
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--spatyper_repeats | string | Lista de repetições spa | |
--spatyper_repeat_order | string | Lista de tipos spa e ordem das repetições | |
--spatyper_do_enrich | boolean | false | Realizar enriquecimento de produto de PCR |
Parâmetros do sccmec
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--sccmec_min_targets_pident | integer | 90 | Porcentagem mínima de identidade para contar um hit de alvo |
--sccmec_min_targets_coverage | integer | 80 | Porcentagem mínima de cobertura para contar um hit de alvo |
--sccmec_min_regions_pident | integer | 85 | Porcentagem mínima de identidade para contar um hit de região |
--sccmec_min_regions_coverage | integer | 93 | Porcentagem mínima de cobertura para contar um hit de região |
Parâmetros do STECFinder
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--stecfinder_use_reads | boolean | false | Reads paired-end do Illumina serão usados em vez de montagens |
--stecfinder_hits | boolean | false | Mostrar resultados detalhados de busca de genes |
--stecfinder_cutoff | number | 10.0 | Cobertura mínima de reads para o gene ser chamado |
--stecfinder_length | number | 50.0 | Porcentagem do comprimento do gene necessária para chamada positiva |
--stecfinder_ipah_length | number | 10.0 | Porcentagem do comprimento do gene ipaH necessária para chamada positiva |
--stecfinder_ipah_depth | number | 1.0 | Profundidade mínima para chamada positiva do gene ipaH (requer --stecfinder_use_reads) |
--stecfinder_stx_length | number | 10.0 | Porcentagem do comprimento do gene stx necessária para chamada positiva |
--stecfinder_stx_depth | number | 1.0 | Profundidade mínima para chamada positiva do gene stx (requer --stecfinder_use_reads) |
--stecfinder_o_length | number | 60.0 | Porcentagem do comprimento do gene wz_ necessária para chamada positiva |
--stecfinder_o_depth | number | 1.0 | Profundidade mínima para chamada positiva do gene qz_ (requer --stecfinder_use_reads) |
--stecfinder_h_length | number | 60.0 | Porcentagem do comprimento do gene fliC necessária para chamada positiva |
--stecfinder_h_depth | number | 1.0 | Profundidade mínima para chamada positiva do gene fliC (requer --stecfinder_use_reads) |
Parâmetros do TB-Profiler Profile
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--tbprofiler_call_whole_genome | boolean | false | Chamar o genoma completo |
--tbprofiler_mapper | string | bwa | Ferramenta de mapeamento a usar. Se estiver usando dados de nanopore, o padrão será minimap2 (opções: bwa, minimap2, bowtie2, bwa-mem2) |
--tbprofiler_caller | string | freebayes | Ferramenta de chamada de variantes a usar (opções: bcftools, gatk, freebayes) |
--tbprofiler_calling_params | string | Opções extras do chamador de variantes entre aspas | |
--tbprofiler_suspect | boolean | false | Usar o conjunto de ferramentas suspect para adicionar predições de ML |
--tbprofiler_no_flagstat | boolean | false | Não coletar flagstats |
--tbprofiler_no_delly | boolean | false | Não executar delly |
--tbprofiler_opts | string | Opções extras entre aspas para o TBProfiler |
Parâmetros do TB-Profiler Collate
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--tbprofiler_itol | boolean | false | Gerar arquivos de configuração do iTOL |
--tbprofiler_full | boolean | false | Gerar mutações no arquivo de resultado principal |
--tbprofiler_all_variants | boolean | false | Gerar todas as variantes na matriz de variantes |
--tbprofiler_mark_missing | boolean | false | Um asterisco será usado para marcar predições afetadas por dados ausentes em uma posição de resistência a drogas |
Parâmetros de Dataset
Define onde o pipeline deve encontrar os dados de entrada e salvar os dados de saída.
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--species | string | Nome da espécie para usar o conjunto de dados específico da espécie | |
--ask_merlin | boolean | Pedir ao Merlin para executar ferramentas Bactopia específicas por espécie com base nas distâncias Mash | |
--coverage | integer | 100 | Reduzir amostras a uma cobertura específica, requer o tamanho do genoma |
--genome_size | integer | 0 | Tamanho esperado do genoma (bp) para todas as amostras, necessário para correção de erros e subamostragem de reads |
--use_bakta | boolean | Usar Bakta para anotação, em vez de Prokka |
Parâmetros Opcionais
Estes parâmetros opcionais podem ser úteis em determinadas situações.
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--outdir | string | bactopia | Diretório base para gravar os resultados |
--skip_compression | boolean | false | Os arquivos de saída não serão comprimidos |
--datasets | string | O caminho para armazenar em cache os conjuntos de dados | |
--keep_all_files | boolean | false | Mantém todos os arquivos de análise criados |
Parâmetros de Requisição Máxima de Jobs
Define o limite máximo de recursos solicitados para qualquer job individual.
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--max_retry | integer | 3 | Número máximo de tentativas de um processo antes de permitir que ele falhe |
--max_cpus | integer | 4 | Número máximo de CPUs que podem ser solicitadas para qualquer job individual |
--max_memory | string | 128.GB | Quantidade máxima de memória que pode ser solicitada para qualquer job individual |
--max_time | string | 240.h | Quantidade máxima de tempo que pode ser solicitada para qualquer job individual |
--max_downloads | integer | 3 | Número máximo de amostras a baixar ao mesmo tempo |
Parâmetros de Configuração do Nextflow
Parâmetros para ajustar a configuração do Nextflow.
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--nfconfig | string | Um arquivo de configuração compatível com Nextflow para perfis personalizados, carregado por último e que substituirá variáveis existentes se definido | |
--publish_dir_mode | string | copy | Método usado para salvar os resultados do pipeline no diretório de saída (opções: symlink, rellink, link, copy, copyNoFollow, move) |
--infodir | string | ${params.outdir}/pipeline_info | Diretório para manter logs e relatórios do Nextflow do pipeline |
--force | boolean | false | O Nextflow sobrescreverá arquivos de saída existentes |
--cleanup_workdir | boolean | false | Após a execução bem-sucedida do Bactopia, o diretório work será excluído |
Opções de configuração institucional
Parâmetros usados para descrever perfis de configuração centralizados. Estes não devem ser editados.
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--custom_config_version | string | master | ID de commit Git para configurações institucionais |
--custom_config_base | string | https://raw.githubusercontent.com/nf-core/configs/master | Diretório base para configurações institucionais |
--config_profile_name | string | Nome da configuração institucional | |
--config_profile_description | string | Descrição da configuração institucional | |
--config_profile_contact | string | Informações de contato da configuração institucional | |
--config_profile_url | string | URL da configuração institucional |
Parâmetros de Perfil do Nextflow
Parâmetros para ajustar a configuração do Nextflow.
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--condadir | string | Diretório que o Nextflow deve usar para ambientes Conda | |
--registry | string | quay.io | Registro para baixar contêineres Docker |
--datasets_cache | string | <HOME>/.bactopia/datasets | Diretório onde os conjuntos de dados baixados devem ser armazenados |
--singularity_cache | string | Diretório onde imagens Singularity remotas são armazenadas | |
--singularity_pull_docker_container | boolean | Em vez de baixar imagens Singularity diretamente, forçar o fluxo de trabalho a baixar e converter contêineres Docker | |
--force_rebuild | boolean | false | Forçar a sobrescrita de ambientes pré-compilados existentes |
--queue | string | general,high-memory | Nome(s) da(s) fila(s) separado(s) por vírgula a serem usados por um agendador de jobs (ex: AWS Batch ou SLURM) |
--cluster_opts | string | Opções adicionais a passar ao executor (ex: SLURM: '--account=my_acct_name') | |
--container_opts | string | Opções adicionais a passar ao Apptainer, Docker ou Singularity (ex: Singularity: '-D pwd') | |
--disable_scratch | boolean | false | Todos os arquivos intermediários criados nos nós de trabalho serão transferidos para o nó principal |
Parâmetros Úteis
Parâmetros raramente usados que podem ser úteis.
| Parâmetro | Tipo | Padrão | Descrição |
|---|---|---|---|
--monochrome_logs | boolean | Não usar saídas de log coloridas | |
--nfdir | boolean | Imprimir o diretório para o qual o Nextflow baixou o Bactopia | |
--sleep_time | integer | 5 | O tempo (em segundos) que o Nextflow aguardará após configurar os conjuntos de dados antes da execução |
--validate_params | boolean | true | Booleano para validar parâmetros em relação ao esquema em tempo de execução |
--help | boolean | Exibir texto de ajuda | |
--wf | string | bactopia | Especificar qual fluxo de trabalho ou Bactopia Tool executar |
--list_wfs | boolean | Listar os fluxos de trabalho e Bactopia Tools disponíveis para usar com '--wf' | |
--show_hidden_params | boolean | Mostrar todos os parâmetros ao usar --help | |
--help_all | boolean | Um alias para --help --show_hidden_params | |
--version | boolean | Exibir texto de versão |
Composição
Este fluxo de trabalho usa os seguintes subworkflows:
- amrfinderplus - Encontrar genes de resistência antimicrobiana e mutações pontuais.
- bactopia_assembler - Montar genomas bacterianos usando seleção automatizada de montador.
- bactopia_datasets - Baixar e fornecer conjuntos de dados pré-compilados necessários pelo Bactopia.
- bactopia_gather - Buscar, validar, reunir e padronizar amostras de entrada.
- bactopia_qc - Realizar controle de qualidade abrangente em reads de sequenciamento.
- bactopia_sketcher - Criar esboços genômicos e realizar classificação taxonômica rápida.
- bakta - Anotação rápida de genomas bacterianos.
- merlin - MinER assisted species-specific bactopia tool seLectIoN.
- mlst - Determinar tipos de sequência multilocus (MLST) a partir de montagens bacterianas.
- prokka - Anotar genomas bacterianos com informações funcionais.