Guia para Iniciantes
Bactopia é um pipeline completo para análise de genomas bacterianos, que inclui mais de 150 ferramentas de bioinformática. Nesta seção, vamos discutir os parâmetros mais essenciais que os usuários precisarão usar para começar com o Bactopia. Vamos nos concentrar nos parâmetros associados ao processamento de amostras de entrada.
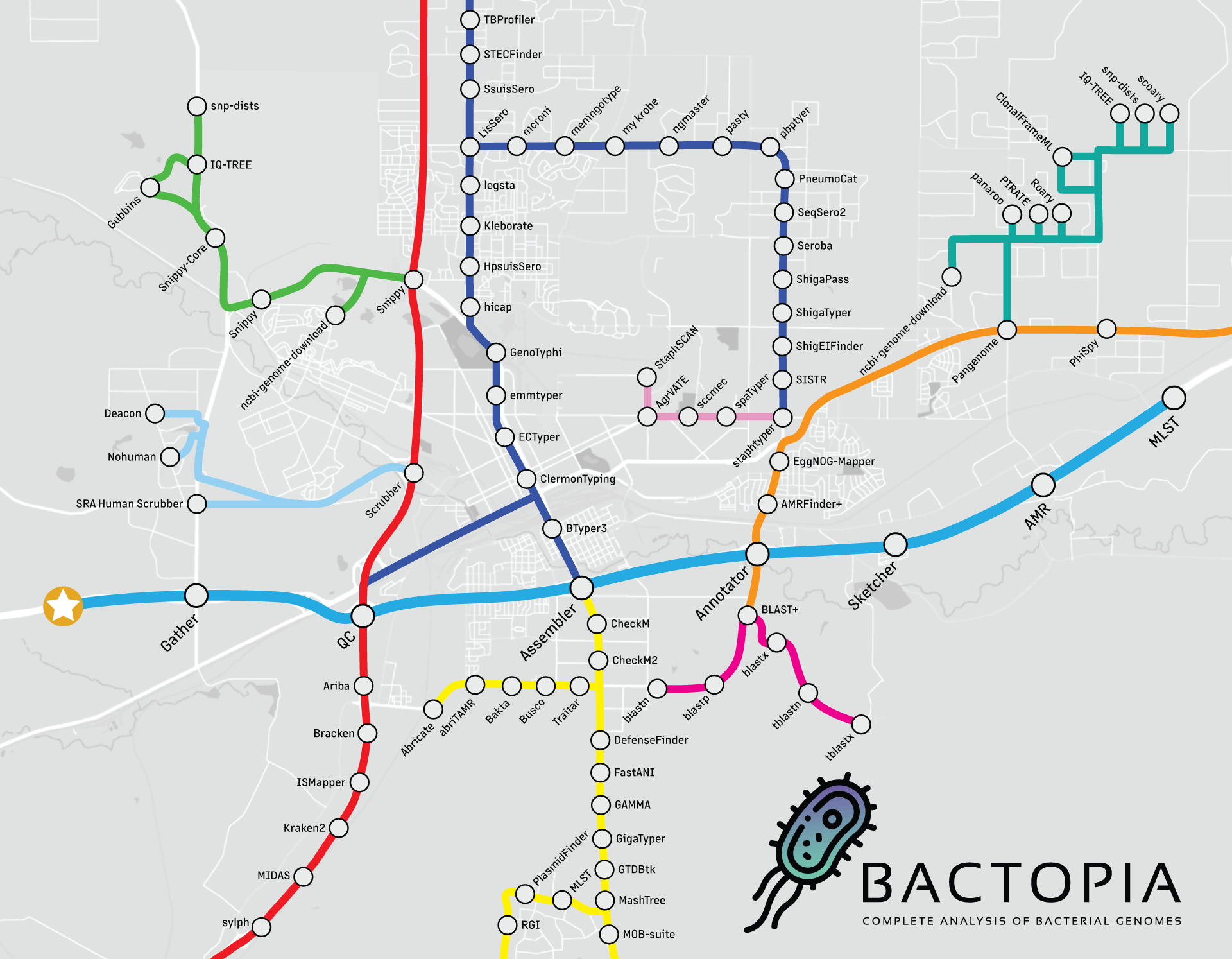
Observando a visão geral do fluxo de trabalho abaixo, vamos realmente nos concentrar no primeiro passo, o passo Gather. Este passo reúne todos os dados em um único lugar, sejam FASTQs locais, FASTQs do SRA ou ENA, ou montagens do NCBI Assembly. O guia a seguir fornecerá alguns exemplos de cada uma dessas entradas aceitas, incluindo:
- Reads Illumina e/ou Nanopore locais
- Montagens locais
- Accessions de Experimentos ENA/SRA
- Accessions do NCBI Assembly
Ao final deste guia, também vamos dar uma olhada em alguns parâmetros úteis. Se você estiver interessado em aprender mais sobre o conjunto completo de parâmetros disponíveis no Bactopia, consulte a seção Guia Completo.
Coletando Entradas
Abaixo está uma tabela com os parâmetros essenciais que você precisará para começar a usar o Bactopia. Isso não significa que você precisa usá-los todos de uma vez, mas será útil se familiarizar com eles. Vamos começar aqui, com uma breve descrição de cada parâmetro, e depois entraremos em mais detalhes sobre cada um com exemplos de casos de uso.
Parâmetros de Entrada
Os parâmetros a seguir são como você fornecerá amostras locais ou remotas para serem processadas pelo Bactopia.
| Parâmetro | Descrição |
|---|---|
--samples | Um FOFN (via bactopia prepare) com nomes de amostras e caminhos para FASTQ/FASTAs a serem processados Tipo: string |
--r1 | Primeiro conjunto de reads Illumina paired-end comprimidos (gzip) (requer --r2 e --sample) Tipo: string |
--r2 | Segundo conjunto de reads Illumina paired-end comprimidos (gzip) (requer --r1 e --sample) Tipo: string |
--se | Reads Illumina single-end comprimidos (gzip) (requer --sample) Tipo: string |
--ont | Reads Oxford Nanopore comprimidos (gzip) (requer --sample) Tipo: string |
--hybrid | Cria montagem híbrida usando Unicycler. (requer --r1, --r2, --ont e --sample) Tipo: boolean |
--short_polish | Cria montagem híbrida a partir de montagem long-read e polimento com short reads. (requer --r1, --r2, --ont e --sample) Tipo: boolean |
--sample | Nome da amostra a ser usado para as sequências de entrada Tipo: string |
--accessions | Um arquivo contendo accessions de Experimentos ENA/SRA ou accessions do NCBI Assembly a serem processados Tipo: string |
--accession | Nome da amostra a ser usado para as sequências de entrada Tipo: string |
--assembly | Um genoma montado em formato FASTA comprimido. (requer --sample) Tipo: string |
--check_samples | Valida o FOFN de entrada fornecido por --samples Tipo: boolean |
Agora vamos dar uma olhada em cada parâmetro com mais detalhes e alguns exemplos de casos de uso.
Amostra Única
Não é segredo que o Bactopia aceita muitos tipos diferentes de entradas a partir de um único ponto de entrada (ou seja, você não precisa de um pipeline separado para cada tipo de entrada). Por enquanto, vamos olhar para entradas locais. Em outras palavras, entradas que já estão na máquina em que você executará o Bactopia. Vamos analisar as seguintes entradas:
- Reads Illumina e/ou Nanopore locais
- Montagens locais
- Processamento de Múltiplas Amostras
Reads Illumina e/ou Nanopore
Vamos começar com as entradas mais comuns: arquivos FASTQ simples para uma amostra única. O Bactopia aceita reads Illumina (paired-end ou single-end) e Nanopore, e pode até processá-los juntos para uma montagem híbrida.
Novamente, aqui estamos focando no processamento de uma única amostra por vez. Para fazer
isso, você deve fornecer uma combinação do nome da amostra (--sample) e o tipo de entrada:
| Tipo de Entrada | Parâmetros Necessários |
|---|---|
| Illumina Paired-End | --r1 e --r2 |
| Illumina Single-End | --se |
| Oxford Nanopore | --ont |
| Híbrido | --r1, --r2, --ont e --hybrid |
| Híbrido (Polimento Short-read) | --r1, --r2, --ont e --short_polish |
--sample é sempre obrigatório para processamento de amostra únicaAo processar uma única amostra, você sempre terá que fornecer --sample,
independentemente do tipo de entrada. Este parâmetro é usado para nomear os arquivos
e diretórios de saída.
Paired-End
Neste exemplo, o Bactopia processará a amostra como reads Illumina paired-end. Os
parâmetros --r1 e --r2 são usados para especificar o local do primeiro e segundo
par de reads. Além disso, o valor de --sample será usado como prefixo
(ex.: my-sample.fna.gz) para salvar os resultados.
bactopia \
--sample my-sample \
--r1 /path/to/my-sample_R1.fastq.gz \
--r2 /path/to/my-sample_R2.fastq.gz
Single-End
Neste exemplo, o Bactopia processará a amostra como reads Illumina single-end. O
parâmetro --se é usado para especificar o local dos reads single-end. Novamente, o
valor de --sample será usado como prefixo para salvar os resultados.
bactopia \
--sample my-sample \
--se /path/to/my-sample.fastq.gz
Nanopore
Vamos mudar um pouco o ritmo: para processar reads Nanopore você precisará de --ont
para especificar o local dos reads Nanopore, bem como --sample para nomear as saídas.
bactopia \
--sample my-sample \
--ont /path/to/my-sample.fastq.gz
Montagem Híbrida
Agora estamos começando a entrar na parte interessante! Digamos que você tenha reads
Illumina paired-end e reads Nanopore para uma amostra. Você pode usar o Bactopia para
criar uma montagem híbrida usando ambos os conjuntos de reads. Para fazer isso, você
passará os reads usando --r1, --r2 (para reads Illumina) e --ont (para reads
Nanopore). Junto com esses, você também fornecerá o parâmetro --hybrid, que dirá ao
Bactopia para criar uma montagem híbrida usando
Unicycler, que monta os short-reads primeiro e
depois preenche as lacunas com os long-reads.
bactopia \
--sample my-sample \
--r1 /path/to/my-sample_R1.fastq.gz \
--r2 /path/to/my-sample_R2.fastq.gz \
--ont /path/to/my-sample.fastq.gz \
--hybrid
Montagem Híbrida (Polimento com Short-read)
Muito similar ao --hybrid, você passará os reads usando --r1, --r2 (para reads
Illumina) e --ont (para reads Nanopore). Desta vez, porém, você usará --short_polish,
que dirá ao Bactopia para criar uma montagem híbrida usando
Dragonflye para montar os long-reads primeiro
e depois realizar o polimento com os short-reads.
bactopia \
--sample my-sample \
--r1 /path/to/my-sample_R1.fastq.gz \
--r2 /path/to/my-sample_R2.fastq.gz \
--ont /path/to/my-sample.fastq.gz \
--short_polish
--short_polish em vez de --hybrid com sequenciamento ONT recenteUsar o Unicycler (--hybrid) para criar uma
montagem híbrida funciona muito bem quando você tem long-reads com baixa cobertura e
muito ruído. No entanto, se você estiver usando sequenciamento ONT recente, provavelmente
tem alta cobertura e usar o método --short_polish vai produzir resultados melhores
(e ser mais rápido!) do que --hybrid.
Bem! Estas são todas as maneiras de processar seus reads Illumina e/ou Nanopore locais. Agora, vamos para as montagens!
Montagem
Vamos imaginar que, por algum motivo, você não tem acesso aos reads brutos de uma amostra,
apenas à montagem. Isso acontece, mas o Bactopia tem uma solução! Você pode usar o
parâmetro --assembly para dizer ao Bactopia para usar a montagem nas análises
downstream.
Quando você fornece uma montagem, algumas coisas acontecem.
- As montagens terão reads Illumina 2x250bp simulados sem inserções ou deleções na sequência e uma pontuação PHRED mínima de Q33.
- Por padrão, a montagem de entrada será usada para todas as análises downstream
(ex.: anotação) que utilizam uma montagem. Caso contrário, se o parâmetro
--reassemblefor fornecido, uma montagem será criada a partir dos reads simulados.
bactopia \
--sample my-sample \
--assembly /path/to/my-sample.fna.gz
Accession ENA/SRA
O predecessor do Bactopia, Staphopia, dependia muito da capacidade de acessar FASTQs disponíveis publicamente no European Nucleotide Archive (ENA) e no Sequence Read Archive (SRA). Era importante que essa capacidade de acessar rapidamente milhões de amostras fosse mantida no Bactopia.
Então, se você quiser incluir amostras disponíveis publicamente em sua análise, o Bactopia
tem isso integrado! Você pode fornecer um accession de Experimento (--accession), e o
Bactopia usará fastq-dl para baixar
automaticamente os arquivos FASTQ associados do ENA ou SRA. Em seguida, o arquivo FASTQ
baixado será processado pelo Bactopia exatamente como seus FASTQs locais normais.
bactopia \
--accession SRX000000
Na hierarquia de accessions, os accessions de Experimento são realmente os únicos únicos. Por exemplo, múltiplos accessions de Run podem estar associados a um único accession de Experimento. Ou, múltiplos accessions de Experimento podem estar associados a um único accession de BioSample. Portanto, ao usar accessions de Experimento, você pode ter certeza de que está obtendo apenas as sequências associadas a esse Experimento "único".
Não é problema nenhum! Você pode usar bactopia search para encontrar rapidamente
quaisquer accessions de Experimento associados ao seu accession. Consulte os exemplos
abaixo para mais informações.
Nos casos em que um único Experimento pode ter múltiplos accessions de Run associados a ele, os arquivos FASTQ de cada Run são mesclados em um único conjunto de sequências.
Accession do NCBI Assembly
Se você pode processar montagens e baixar FASTQs do ENA/SRA de forma transparente, faz
sentido que você também possa processar montagens do NCBI Assembly! Similar ao download
de FASTQs do ENA/SRA, você pode fornecer um accession do NCBI Assembly usando
--accession. Esses accessions são os que começam com GCF ou GCA. Quando fornecido
um accession do NCBI Assembly, o Bactopia usará
ncbi-genome-download para buscar a
montagem associada e processá-la como uma montagem local.
bactopia \
--accession GCF_000000000
Com o tempo, descobri que a versão da montagem pode ser instável. Por exemplo, às vezes uma montagem pode ser corrigida e a versão anterior deixa de estar disponível. Portanto, para evitar problemas, o Bactopia sempre usará a versão mais recente de um dado accession do NCBI Assembly.
Múltiplas Amostras
A esta altura, você já deve ter uma boa compreensão de como processar uma única amostra, mas pode estar pensando: "Tenho centenas de amostras, não quero executar o Bactopia centenas de vezes. Posso executar todas de uma vez?" A resposta é SIM!
O Bactopia permite que você forneça um arquivo de nomes de arquivos (FOFN) usando
--samples ou uma lista de accessions usando --accessions. Usando qualquer um desses
parâmetros, você pode processar uma única ou
milhares de amostras
em um único comando.
Nesta seção, vamos ver como processar múltiplas amostras usando --samples e
--accessions. Também veremos os comandos auxiliares do Bactopia para ajudar a gerar
o FOFN ou a lista de accessions adequados para processar múltiplas amostras.
Aqui está uma pequena tabela para ajudar você a decidir qual parâmetro usar:
| Parâmetro | Aplicação | Comando Auxiliar |
|---|---|---|
--samples | Amostras Locais | bactopia prepare |
--accessions | Accessions ENA/SRA e Assembly | bactopia search |
Agora, vamos entrar em mais detalhes sobre cada um deles.
Amostras Locais
Acima, você aprendeu como usar parâmetros como --r1 e --r2 para processar uma única
amostra, mas você tem muitas amostras para as quais acabou de receber sequências. Agora
você planeja executar o Bactopia em cada amostra, então vamos aprender como gerar um
FOFN, ou samplesheet, que você pode passar para o Bactopia para processar todas as
amostras de uma vez.
Primeiro, mencionei arquivo de nomes de arquivos (FOFN) algumas vezes, mas o que é isso? Um FOFN é um arquivo que contém uma lista de amostras e seus FASTQs/FASTAs associados. Um arquivo, de nomes de arquivos.
Para o Bactopia, esse FOFN é uma tabela delimitada por tabulação com cinco colunas:
| Coluna | Descrição |
|---|---|
| sample | Um prefixo único, ou nome único, a ser usado para nomear os arquivos de saída |
| runtype | Informa ao Bactopia que tipo de entrada a amostra é (ex.: paired-end, single-end, nanopore, etc...) |
| genome_size | O tamanho de genoma esperado para a amostra fornecida |
| species | A classificação taxonômica esperada para a amostra fornecida |
| r1 | Se paired-end, o primeiro par de reads; caso contrário, os reads single-end |
| r2 | Se paired-end, o segundo par de reads |
| extra | A montagem ou long reads associados a uma amostra |
Com isso em mente, vamos ver um exemplo de FOFN:
sample runtype genome_size species r1 r2 extra
s01 paired-end 180000 Bacterial species /fq/s01_R1_001.fastq.gz /fq/s01_R2_001.fastq.gz
s02 paired-end 180000 Bacterial species /fq/s02_R1_001.fastq.gz /fq/s02_R2_001.fastq.gz
s03 single-end 180000 Bacterial species /fq/s03_001.fastq.gz
Com este FOFN, você pode usar --samples para processar todas as três amostras de uma vez.
Usar --samples pode economizar muito tempo para você, e é sempre recomendado
adotar essa abordagem quando possível.
bactopia \
--samples my-samples.txt
Agora, você pode estar pensando: "Não quero criar um FOFN manualmente, isso dá muito trabalho!"
Ótima notícia! O Bactopia tem um comando auxiliar integrado para ajudá-lo a gerar um FOFN
automaticamente. Vamos dar uma olhada em bactopia prepare.
bactopia prepare
Embora seja possível criar manualmente o FOFN necessário, não é recomendado. Pode ser
um pouco tedioso e sujeito a erros, portanto, evite criar seu FOFN manualmente. Em vez
disso, use bactopia prepare para gerar com precisão um FOFN para suas amostras.
Quando o Bactopia recebe um FOFN, a primeira coisa que ele faz é verificar se todos os arquivos de entrada foram encontrados e comprimidos usando Gzip. Se tudo estiver correto, cada amostra será processada; caso contrário, uma lista de amostras com erros será enviada para o STDERR.
Use `--check_samples` para validar apenas o FOFN
Se você quiser apenas validar seu FOFN (e não executar o pipeline completo), você pode
usar o parâmetro --check_samples. No entanto, se você usou bactopia prepare para
gerar seu FOFN, ele deve ser válido.
Honestamente, bactopia prepare é uma daquelas ferramentas que é melhor explicada com
exemplos. Então, vamos dar uma olhada em alguns exemplos.
Exemplos
Usar bactopia prepare pode ser um pouco complicado no início, mas depois que você
pegar o jeito, vai se descobrir usando-o o tempo todo.
bactopia prepare usa por padrão <SAMPLE_NAME>_R1.fastq.gz e <SAMPLE_NAME>_R2.fastq.gz
para reads paired-end, e <SAMPLE_NAME>.fastq.gz para reads single-end. Usar nomes de
arquivo que sigam esse padrão vai ajudá-lo a evitar o uso de expressões regulares.
bactopia prepare deve conseguir lidar com sua configuração para gerar o FOFN adequado,
mas pode ser necessário algum esforço. Vamos ver os parâmetros disponíveis e alguns exemplos.
Parâmetros disponíveis do `bactopia prepare`
| Parâmetro | Descrição |
|---|---|
--path | O diretório onde seus FASTQs/FASTAs estão armazenados. |
--assembly_ext | A extensão dos seus FASTAs. Padrão: .fna.gz |
--fastq_ext | A extensão dos seus FASTQs. Padrão: .fastq.gz |
--fastq_separator | O caractere usado para dividir o nome do FASTQ. Padrão: _ |
--pe1_pattern | A expressão regular para corresponder ao primeiro par de reads paired-end. Padrão: ([Aa]|[Rr]1|1) |
--pe2_pattern | A expressão regular para corresponder ao segundo par de reads paired-end. Padrão: ([Bb]|[Rr]2|2) |
--merge | Sinaliza amostras com múltiplos conjuntos de reads para serem mescladas pelo Bactopia. |
--ont | Sinaliza reads single-end para serem tratados como reads Oxford Nanopore. |
--recursive | Sinaliza a busca recursiva de diretórios por FASTQs/FASTAs. |
--prefix | Substitui o caminho absoluto por uma string fornecida. Padrão: Usar caminho absoluto |
--metadata | Metadados por amostra com informações de tamanho de genoma e espécie. |
--genome-size | Tamanho de genoma a ser usado para todas as amostras. |
--species | Espécie a ser usada para todas as amostras (se disponível, pode ser usada para determinar o tamanho do genoma). |
--taxid | Usa o tamanho do genoma do ID de Taxon para todas as amostras. |
Reads Illumina
Digamos que você tenha um diretório de reads Illumina paired-end. Os arquivos são
nomeados para corresponder às expectativas padrão: <SAMPLE_NAME>_R1.fastq.gz,
<SAMPLE_NAME>_R2.fastq.gz e <SAMPLE_NAME>.fastq.gz. Você pode usar
bactopia prepare para gerar um FOFN para você.
bactopia prepare --path /path/to/fastqs
Isso vai gerar um FOFN que se parece com isso:
sample runtype genome_size species r1 r2 extra
s01 paired-end 180000 unknown /path/to/fastqs/s01_R1.fastq.gz /path/to/fastqs/s01_R2.fastq.gz
s02 paired-end 180000 unknown /path/to/fastqs/s02_R1.fastq.gz /path/to/fastqs/s02_R2.fastq.gz
s03 single-end 180000 unknown /path/to/fastqs/s03.fastq.gz
Reads Oxford Nanopore
Digamos que você tenha um diretório de reads Oxford Nanopore. Os arquivos são nomeados
para corresponder às expectativas padrão: <SAMPLE_NAME>.fastq.gz. Você pode usar
bactopia prepare para gerar um FOFN para você.
bactopia prepare --path /path/to/fastqs --ont
Ao usar --ont, quaisquer reads single-end encontrados serão tratados como reads ONT.
Isso vai gerar um FOFN que se parece com isso:
sample runtype genome_size species r1 r2 extra
s03 ont 180000 unknown /path/to/fastqs/s01.fastq.gz
Reads Illumina Paired-End e Oxford Nanopore
Digamos que você tenha um diretório de reads Illumina paired-end e reads Oxford Nanopore.
Novamente, eles são nomeados para corresponder às expectativas padrão:
<SAMPLE_NAME>_R1.fastq.gz, <SAMPLE_NAME>_R2.fastq.gz e <SAMPLE_NAME>.fastq.gz.
Você pode usar bactopia prepare para gerar um FOFN para você.
bactopia prepare --path /path/to/fastqs --ont
Novamente, use --ont para dizer ao bactopia prepare para tratar quaisquer reads
single-end como reads ONT. Isso vai gerar um FOFN que se parece com isso:
sample runtype genome_size species r1 r2 extra
s01 paired-end 180000 unknown /path/to/fastqs/s01_R1.fastq.gz /path/to/fastqs/s01_R2.fastq.gz
s02 paired-end 180000 unknown /path/to/fastqs/s02_R1.fastq.gz /path/to/fastqs/s02_R2.fastq.gz
s03 ont 180000 unknown /path/to/fastqs/s03.fastq.gz
Mesclando Múltiplos Runs Illumina
Digamos que você tenha um diretório de reads Illumina, mas tem múltiplos runs para cada
amostra e quer que o Bactopia mescle os reads. Novamente, assumindo que eles são nomeados
para corresponder às expectativas padrão: <SAMPLE_NAME>_R1.fastq.gz,
<SAMPLE_NAME>_R2.fastq.gz e <SAMPLE_NAME>.fastq.gz. Você pode usar
bactopia prepare para gerar um FOFN para você.
bactopia prepare --path /path/to/fastqs --merge
Ao usar --merge, quaisquer amostras com múltiplos runs serão mescladas em um único
conjunto de reads. Isso vai gerar um FOFN que se parece com isso:
sample runtype genome_size species r1 r2 extra
s01 merge-pe 180000 unknown /run1/s01_R1.fastq.gz,/run2/s01_R1.fastq.gz /run1/s01_R2.fastq.gz,/run2/s01_R2.fastq.gz
s02 merge-se 180000 unknown /run1/s02.fastq.gz,/run2/s02.fastq.gz
Reads com nomes '*_001.fastq.gz'
Digamos que você tenha um diretório de reads Illumina, mas eles são nomeados com
*_001.fastq.gz em vez das expectativas padrão: <SAMPLE_NAME>_R1.fastq.gz,
<SAMPLE_NAME>_R2.fastq.gz e <SAMPLE_NAME>.fastq.gz. Você pode usar
bactopia prepare, mas precisará fornecer alguns parâmetros adicionais para gerar um
FOFN para você.
bactopia prepare --path /path/to/fastqs --fastq-ext '_001.fastq.gz'
Aqui você precisará usar --fastq-ext para dizer ao bactopia prepare para procurar
*_001.fastq.gz em vez do padrão *.fastq.gz. Isso vai gerar um FOFN que se parece
com isso:
sample runtype genome_size species r1 r2 extra
s01 paired-end 180000 unknown /path/to/fastqs/s01_R1_001.fastq.gz /path/to/fastqs/s01_R2_002.fastq.gz
s02 paired-end 180000 unknown /path/to/fastqs/s02_R1_001.fastq.gz /path/to/fastqs/s02_R2_002.fastq.gz
s03 single-end 180000 unknown /path/to/fastqs/s03_001.fastq.gz
Há muitas combinações possíveis de parâmetros que você pode usar com bactopia prepare.
Se você estiver travado em alguma ou quiser ver um exemplo, por favor me avise
Abrindo uma Issue no GitHub.
Accessions
Se você começou do início e chegou até aqui, parabéns! De qualquer forma, acima você
aprendeu que pode usar --accession para baixar FASTQs do ENA/SRA ou montagens do NCBI
Assembly. Depois, aprendeu que pode usar --samples para processar quantas amostras
quiser. Então, faz sentido que exista um complemento para --samples para processar
múltiplos accessions de uma vez! Esse parâmetro é --accessions.
Usar --accessions pode economizar muito tempo para você, permitindo que você processe
quantos genomas disponíveis publicamente quiser.
bactopia \
--accessions my-accessions.txt
De forma similar ao --samples, há um comando auxiliar complementar chamado
bactopia search que permite enviar uma consulta e gerar uma lista de accessions de
Experimento a serem processados pelo Bactopia (via --accessions).
Vamos dar uma olhada em bactopia search e como ele pode te ajudar.
bactopia search
bactopia search foi criado para ajudar a gerar uma lista de accessions de Experimento
a serem processados pelo Bactopia (via --accessions). Você pode fornecer um ID de Taxon
(ex.: 1280), um nome de organismo (ex.: Staphylococcus aureus), um accession de Study
(ex.: PRJNA480016), um accession de BioSample (ex.: SAMN01737350) ou um accession de Run
(ex.: SRR578340). Esse valor é então consultado na
Data Warehouse API) do ENA, e uma lista
de todos os accessions de Experimento associados à consulta é retornada.
Novamente, provavelmente é mais fácil se simplesmente olharmos alguns exemplos.
Exemplos
Primeiro, vamos ver um único exemplo para fornecer uma descrição dos arquivos de saída.
bactopia search --query PRJNA480016 --limit 5
INFO 2023 00:root:INFO - Submitting query (type - bioproject_accession) search.py:472
INFO 2023 00:root:INFO - Writing results to ./bactopia-metadata.txt search.py:554
INFO 2023 00:root:INFO - Writing accessions to ./bactopia-accessions.txt search.py:564
INFO 2023 00:root:INFO - Writing filtered accessions to ./bactopia-filtered.txt search.py:569
INFO 2023 00:root:INFO - Writing summary to ./bactopia-search.txt search.py:575
No comando acima, estamos buscando todos os accessions de Experimento associados ao
accession de Study PRJNA480016. No entanto, o parâmetro --limit é usado para limitar
os resultados a apenas 5 accessions de Experimento. Em seguida, múltiplos arquivos são
produzidos:
| Extensão | Descrição |
|---|---|
-metadata.txt | Um arquivo delimitado por tabulação com todos os resultados da consulta |
-accessions.txt | Uma lista de accessions de Experimento a serem processados |
-filtered.txt | Uma lista de quaisquer accessions de Experimento que foram filtrados, caso contrário vazio |
-search.txt | Um resumo da solicitação concluída |
Exemplo bactopia-metadata.txt
Quando concluído, um arquivo chamado bactopia-metadata.txt é produzido. Este arquivo
contém múltiplos campos (sample_accession, tax_id, sample_alias, center_name, etc...)
para cada accession de Experimento retornado pela consulta.
run_accession project_name submission_accession library_selection last_updated sra_bytes collected_by isolate fastq_bytes instrument_platform sra_aspera fastq_galaxy country sample_description experiment_title sra_galaxy fastq_md5 sample_accession secondary_study_accession read_count study_title collection_date_end sample_title instrument_model description sra_md5 fastq_ftp base_count library_strategy location library_source sra_ftp library_layout location_start status lon fastq_aspera host_sex sample_alias collection_date_start run_alias collection_date experiment_alias center_name host library_name tag first_created lat strain experiment_accession scientific_name tax_id study_accession host_scientific_name accession secondary_sample_accession location_end first_public study_alias isolation_source
SRR7706353 Staphylococcus aureus SRA760272 RANDOM 2018-08-18 595393300 353569630;334112090 ILLUMINA fasp.sra.ebi.ac.uk:/vol1/srr/SRR770/003/SRR7706353 ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/003/SRR7706353/SRR7706353_1.fastq.gz;ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/003/SRR7706353/SRR7706353_2.fastq.gz USA Pathogen: clinical or host-associated sample from Staphylococcus aureus Illumina MiSeq sequencing: Genome Sequence of Staphylococcus aureus Cystic Fibrosis Isolates ftp.sra.ebi.ac.uk/vol1/srr/SRR770/003/SRR7706353 6cf7a954abc803c8be6515545b321e2d;f879b1fa058e80fa764beb8e333877ae SAMN09847868 SRP158268 1493115 Genome Sequence of Staphylococcus aureus Cystic Fibrosis Isolates 2023-01-07 Pathogen: clinical or host-associated sample from Staphylococcus aureus Illumina MiSeq Illumina MiSeq sequencing: Genome Sequence of Staphylococcus aureus Cystic Fibrosis Isolates 4f7c2a8836ce2471fec07128f2c9b407 ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/003/SRR7706353/SRR7706353_1.fastq.gz;ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/003/SRR7706353/SRR7706353_2.fastq.gz 897918508 WGS GENOMIC ftp.sra.ebi.ac.uk/vol1/srr/SRR770/003/SRR7706353 PAIRED public fasp.sra.ebi.ac.uk:/vol1/fastq/SRR770/003/SRR7706353/SRR7706353_1.fastq.gz;fasp.sra.ebi.ac.uk:/vol1/fastq/SRR770/003/SRR7706353/SRR7706353_2.fastq.gz JE2 2023-01-07 JE2_R1.fastq.gz 2017-07-01 JE2 SUB4273132 Homo sapiens JE2 ena;pathogen;bacterium;datahub;priority 2018-08-18 JE2 SRX4563690 Staphylococcus aureus 1280 PRJNA480016 Homo sapiens SRR7706353 SRS3680044 2018-08-18 PRJNA480016
SRR7706354 Staphylococcus aureus SRA760272 RANDOM 2018-08-18 227970617 Emory Cystic Fibrosis Biospecimen Registry (CFBR) replicate of CFBRSa66A 129917564;131945147 ILLUMINAfasp.sra.ebi.ac.uk:/vol1/srr/SRR770/004/SRR7706354 ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/004/SRR7706354/SRR7706354_1.fastq.gz;ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/004/SRR7706354/SRR7706354_2.fastq.gz USA: Atlanta, GA MRSA Illumina MiSeq sequencing: Genome Sequence of Staphylococcus aureus Cystic Fibrosis Isolates ftp.sra.ebi.ac.uk/vol1/srr/SRR770/004/SRR7706354 371165d54adfd1300c7b02e79d8d4245;5517e629b8e8ad00dbd6ef9a5f8d073d SAMN09847839 SRP158268 535939 Genome Sequence of Staphylococcus aureus Cystic Fibrosis Isolates 2018-01-02 Pathogen: clinical or host-associated sample from Staphylococcus aureus Illumina MiSeq Illumina MiSeq sequencing: Genome Sequence of Staphylococcus aureus Cystic Fibrosis Isolates 323e7336212b256ba2509a14bd90790a ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/004/SRR7706354/SRR7706354_1.fastq.gz;ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/004/SRR7706354/SRR7706354_2.fastq.gz 322122209 WGS 33.749 N 84.388 W GENOMIC ftp.sra.ebi.ac.uk/vol1/srr/SRR770/004/SRR7706354 PAIRED 33.749 N 84.388 W public -84.388 fasp.sra.ebi.ac.uk:/vol1/fastq/SRR770/004/SRR7706354/SRR7706354_1.fastq.gz;fasp.sra.ebi.ac.uk:/vol1/fastq/SRR770/004/SRR7706354/SRR7706354_2.fastq.gz male CFBRSa66B 2018-01-02 CFBRSa66B_R2.fastq.gz 2012-07-16 CFBRSa66B SUB4273132 Homo sapiens CFBRSa66B ena;pathogen;bacterium;datahub;priority 2018-08-18 33.749 CFBR-150 SRX4563689 Staphylococcus aureus 1280 PRJNA480016 Homo sapiens SRR7706354 SRS3680043 33.749 N 84.388 W 2018-08-18 PRJNA480016 sputum
SRR7706356 Staphylococcus aureus SRA760272 RANDOM 2018-08-18 328642242 Emory Cystic Fibrosis Biospecimen Registry (CFBR) 191742121;188439990 ILLUMINA fasp.sra.ebi.ac.uk:/vol1/srr/SRR770/006/SRR7706356 ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/006/SRR7706356/SRR7706356_1.fastq.gz;ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/006/SRR7706356/SRR7706356_2.fastq.gz USA: Atlanta, GA MRSA Illumina MiSeq sequencing: Genome Sequence of Staphylococcus aureus Cystic Fibrosis Isolates ftp.sra.ebi.ac.uk/vol1/srr/SRR770/006/SRR7706356 2b0c01434a7e677c6697ff49985de0f7;08c4f37d7fdbeac0133819ee3af6dd21 SAMN09847834 SRP158268 780838 Genome Sequence of Staphylococcus aureus Cystic Fibrosis Isolates 2017-09-02 Pathogen: clinical or host-associated sample from Staphylococcus aureus Illumina MiSeq Illumina MiSeq sequencing: Genome Sequence of Staphylococcus aureus Cystic Fibrosis Isolates ec8397df7897777d1c332522c6227458 ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/006/SRR7706356/SRR7706356_1.fastq.gz;ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/006/SRR7706356/SRR7706356_2.fastq.gz 469342822 WGS 33.749 N 84.388 W GENOMIC ftp.sra.ebi.ac.uk/vol1/srr/SRR770/006/SRR7706356 PAIRED 33.749 N 84.388 W public -84.388 fasp.sra.ebi.ac.uk:/vol1/fastq/SRR770/006/SRR7706356/SRR7706356_1.fastq.gz;fasp.sra.ebi.ac.uk:/vol1/fastq/SRR770/006/SRR7706356/SRR7706356_2.fastq.gz male CFBRSa25 2017-09-01 CFBRSa25_R2.fastq.gz 2012-03-26 CFBRSa25 SUB4273132 Homo sapiens CFBRSa25 ena;pathogen;bacterium;datahub;priority 2018-08-18 33.749 CFBR-134 SRX4563687 Staphylococcus aureus 1280 PRJNA480016 Homo sapiens SRR7706356 SRS3680041 33.749 N 84.388 W 2018-08-18 PRJNA480016 sputum
SRR7706361 Staphylococcus aureus SRA760272 RANDOM 2018-08-18 599269367 Emory Cystic Fibrosis Biospecimen Registry (CFBR) 353160072;336993031 ILLUMINA fasp.sra.ebi.ac.uk:/vol1/srr/SRR770/001/SRR7706361 ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/001/SRR7706361/SRR7706361_1.fastq.gz;ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/001/SRR7706361/SRR7706361_2.fastq.gz USA: Atlanta, GA Pathogen: clinical or host-associated sample from Staphylococcus aureus Illumina MiSeq sequencing: Genome Sequence of Staphylococcus aureus Cystic Fibrosis Isolates ftp.sra.ebi.ac.uk/vol1/srr/SRR770/001/SRR7706361 45fa5f0ed629d81282f1429b42c18432;c9c1b6be39fceab54d20d41450776050 SAMN09847850 SRP158268 1496420 Genome Sequence of Staphylococcus aureus Cystic Fibrosis Isolates 2018-04-04 Pathogen: clinical or host-associated sample from Staphylococcus aureus Illumina MiSeq Illumina MiSeq sequencing: Genome Sequence of Staphylococcus aureus Cystic Fibrosis Isolates 085cbb8f7b186d3f61bab022323f61ce ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/001/SRR7706361/SRR7706361_1.fastq.gz;ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/001/SRR7706361/SRR7706361_2.fastq.gz 899864766 WGS 33.749 N 84.388 GENOMIC ftp.sra.ebi.ac.uk/vol1/srr/SRR770/001/SRR7706361 PAIRED 33.749 N 84.388 W public -84.388 fasp.sra.ebi.ac.uk:/vol1/fastq/SRR770/001/SRR7706361/SRR7706361_1.fastq.gz;fasp.sra.ebi.ac.uk:/vol1/fastq/SRR770/001/SRR7706361/SRR7706361_2.fastq.gz male CFBRSa07 2018-04-03 CFBRSa07_R1.fastq.gz 2012-10-03 CFBRSa07 SUB4273132 Homo sapiens CFBRSa07 ena;pathogen;bacterium;datahub;priority 2018-08-18 33.749 CFBR-238 SRX4563682 Staphylococcus aureus 1280 PRJNA480016 Homo sapiens SRR7706361 SRS3680035 33.749 N 84.388 W 2018-08-18 PRJNA480016 sputum
SRR7706362 Staphylococcus aureus SRA760272 RANDOM 2018-08-18 241721284 Emory Cystic Fibrosis Biospecimen Registry (CFBR) 139499004;138853939 ILLUMINA fasp.sra.ebi.ac.uk:/vol1/srr/SRR770/002/SRR7706362 ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/002/SRR7706362/SRR7706362_1.fastq.gz;ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/002/SRR7706362/SRR7706362_2.fastq.gz USA: Atlanta, GA Pathogen: clinical or host-associated sample from Staphylococcus aureus Illumina MiSeq sequencing: Genome Sequence of Staphylococcus aureus Cystic Fibrosis Isolates ftp.sra.ebi.ac.uk/vol1/srr/SRR770/002/SRR7706362 d6f7434e83969245df356e0c3aaa72e8;4bfa95c3a9db9d93b65b39530f5be0c7 SAMN09847844 SRP158268 572961 Genome Sequence of Staphylococcus aureus Cystic Fibrosis Isolates 2017-11-02 Pathogen: clinical or host-associated sample from Staphylococcus aureus Illumina MiSeq Illumina MiSeq sequencing: Genome Sequence of Staphylococcus aureus Cystic Fibrosis Isolates 189012bcc94d59002369ebc17ad303fa ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/002/SRR7706362/SRR7706362_1.fastq.gz;ftp.sra.ebi.ac.uk/vol1/fastq/SRR770/002/SRR7706362/SRR7706362_2.fastq.gz 344400515 WGS 33.749 N 84.388 GENOMIC ftp.sra.ebi.ac.uk/vol1/srr/SRR770/002/SRR7706362 PAIRED 33.749 N 84.388 W public -84.388 fasp.sra.ebi.ac.uk:/vol1/fastq/SRR770/002/SRR7706362/SRR7706362_1.fastq.gz;fasp.sra.ebi.ac.uk:/vol1/fastq/SRR770/002/SRR7706362/SRR7706362_2.fastq.gz male CFBRSa06 2017-11-02 CFBRSa06_R1.fastq.gz 2012-05-16 CFBRSa06 SUB4273132 Homo sapiens CFBRSa06 ena;pathogen;bacterium;datahub;priority 2018-08-18 33.749 CFBR-172 SRX4563681 Staphylococcus aureus 1280 PRJNA480016 Homo sapiens SRR7706362 SRS3680034 33.749 N 84.388 W 2018-08-18 PRJNA480016 sputum
Exemplo bactopia-summary.txt
Quando concluído, um arquivo chamado bactopia-summary.txt é produzido, que conterá um
resumo básico dos resultados da consulta.
Bactopia Summary Report
Total Samples: 1
Passed: 1
Gold: 0
Silver: 1
Bronze: 0
Excluded: 0
Failed Cutoff: 0
QC Failure: 0
Reports:
Full Report (txt): ./bactopia-report.tsv
Exclusion: ./bactopia-exclude.tsv
Summary: ./bactopia-summary.txt
Rank Cutoffs:
Gold:
Coverage >= 100x
Quality >= Q30
Read Length >= 95bp
Total Contigs < 100
Silver:
Coverage >= 50x
Quality >= Q20
Read Length >= 75bp
Total Contigs < 200
Bronze:
Coverage >= 20x
Quality >= Q12
Read Length >= 49bp
Total Contigs < 500
Assembly Length Exclusions:
Minimum: None
Maximum: None
A partir dos arquivos de saída, você vai querer usar o arquivo com a extensão
-accessions.txt. Na consulta acima, o arquivo -accessions.txt ficou assim:
SRX4563681 illumina Staphylococcus aureus 2800000
SRX4563689 illumina Staphylococcus aureus 2800000
SRX4563687 illumina Staphylococcus aureus 2800000
SRX4563682 illumina Staphylococcus aureus 2800000
SRX4563690 illumina Staphylococcus aureus 2800000
-accessions.txt com --accessionsO arquivo com a extensão -accessions.txt é o arquivo que você usará com --accessions
para processar os accessions de Experimento com o Bactopia.
Parâmetros Adicionais Úteis
-profile
O Bactopia faz uso dos Perfis de Configuração do Nextflow
para especificar o executor a ser usado. Por padrão, o Bactopia usará o perfil conda.
Há outros perfis integrados, incluindo: docker, singularity, slurm, etc... Para
usar um perfil específico, você pode usar o parâmetro -profile.
Por exemplo, se você quiser que o Nextflow use Docker, você usaria o seguinte comando:
bactopia ... -profile docker
Com isso, o Nextflow usará Docker para executar todos os processos no Bactopia (mesmo que o Bactopia esteja instalado com Conda!).
Embora eu seja o primeiro a admitir que adoro Conda, ele não é perfeito. Com o tempo, ferramentas podem ficar quebradas ou incompatíveis devido a dependências. Containers são uma ótima maneira de evitar esses problemas. Se você está usando o Bactopia e tem Docker ou Singularity disponível, eu recomendaria usá-los em vez de Conda.
-resume
O Bactopia depende do Recurso de Retomada do Nextflow
para retomar execuções. Você pode dizer ao Bactopia para retomar adicionando -resume
à sua linha de comando. Quando -resume é usado, o Nextflow revisará o cache e
determinará se a execução anterior pode ser retomada. Se a execução anterior não puder
ser retomada, a execução começará do início.
--max_cpus
Na execução, o Nextflow cria uma fila e o número de slots na fila é determinado pelo
número total de núcleos no sistema. Portanto, se você tiver um sistema com 24 núcleos,
isso significa que o Nextflow terá uma fila com 24 slots disponíveis. Esse recurso torna
o --max_cpus um pouco enganoso. Normalmente, quando você fornece --max_cpus, você
está dizendo "use essa quantidade de CPUs". Mas esse não é o caso para o Nextflow e o
Bactopia. Quando você usa --max_cpus, o que você está realmente dizendo é "para
qualquer tarefa específica, use essa quantidade de slots". Os comandos dentro dos
processadores de uma tarefa usarão a quantidade especificada por --max_cpus.
`--max_cpus` pode ter um efeito significativo na eficiência do Bactopia
Por exemplo, se você tiver um sistema com 24 núcleos.
Este comando, bactopia ... --max_cpus 24, diz para qualquer tarefa específica, use
24 slots. O Nextflow dará às tarefas no Bactopia 24 slots de 24 disponíveis (máquina
com 24 núcleos). Em outras palavras, a fila pode ter apenas uma tarefa em execução por
vez porque cada tarefa ocupa 24 slots.
Por outro lado, bactopia ... --max_cpus 4 diz para qualquer tarefa específica, use 4
slots. Agora, o Nextflow dará a cada tarefa 4 slots de 24 slots. Isso significa que 6
tarefas podem estar em execução ao mesmo tempo. Isso pode levar a uma eficiência muito
melhor porque menos tarefas ficam esperando na fila.
Existem algumas tarefas no Bactopia que sempre usarão apenas um único slot porque são
tarefas de núcleo único. Enquanto outras sempre usarão o número de slots especificado
por --max_cpus.
Se o --max_cpus for muito alto, você provavelmente reduzirá a eficiência do Bactopia.
--max_cpus 4 é um valor seguro.Este também é o valor padrão para o Bactopia.
-qs
O parâmetro -qs é a abreviação de queue size (tamanho da fila). Como descrito acima
para --max_cpus, o valor padrão para -qs é definido como o número total de núcleos
no sistema. Este parâmetro permite ajustar o número máximo de núcleos que o Nextflow
pode usar a qualquer momento.
`-qs` permite que você use recursos compartilhados de forma adequada
No exemplo acima, se você tiver um sistema com 24 núcleos, o tamanho padrão da fila é 24 slots.
bactopia ... --max_cpus 4 diz para qualquer tarefa específica, use no máximo 4 slots.
O Nextflow dará a cada tarefa 4 slots de 24 slots. Mas pode haver outras pessoas também
usando o servidor.
bactopia ... --max_cpus 4 -qs 12 diz para qualquer tarefa específica, use no máximo
4 slots, mas não use mais de 12 slots. O Nextflow dará a cada tarefa 4 slots de 12
slots. Agora, em vez de usar todos os núcleos do servidor, o máximo que pode ser usado
é 12.
`-qs` pode precisar de ajuste para agendadores de tarefas.
O valor padrão para -qs é definido como 100 ao usar um agendador de tarefas (ex.:
SLURM, AWS Batch). Pode haver situações em que você precise ajustar isso para atender
às suas necessidades. Por exemplo, ao usar o AWS Batch, você pode querer aumentar o
valor para ter mais tarefas processadas de uma vez (ex.: 100 vs 500).
--genome_size
Ao longo do fluxo de trabalho do Bactopia, um tamanho de genoma é usado para várias tarefas. Por padrão, o tamanho do genoma é definido como 0 e etapas como a redução de cobertura são ignoradas. No entanto, se você fornecer um tamanho de genoma esperado, essas etapas serão habilitadas.
Use `--genome_size` para melhorar os resultados e a velocidade
Ao fornecer um tamanho de genoma, o Bactopia reduzirá a cobertura para um máximo de 100x (padrão). Ao fazer isso, para amostras com cobertura superior a 100x, você verá uma redução no tempo de execução, bem como melhores resultados. Isso porque, com cobertura excessiva, algumas ferramentas produzem resultados piores e levam muito mais tempo.
--nfconfig
Um recurso fundamental do Nextflow é que você pode fornecer seus próprios arquivos de
configuração. Isso basicamente significa que você pode configurar facilmente o Bactopia
para rodar no seu ambiente. Com --nfconfig, você pode dizer ao Bactopia para importar
seu arquivo de configuração.
--nfconfig foi configurado para ser o último arquivo de configuração a ser carregado
pelo Nextflow. Isso significa que, se seu arquivo de configuração contiver variáveis
(ex.: params ou profiles) já definidas, elas serão substituídas pelos seus valores.
O Nextflow entra em grande detalhe sobre como criar arquivos de configuração. Por favor, verifique os links a seguir para os ajustes que você possa estar interessado em fazer.
| Escopo | Descrição |
|---|---|
| env | Definir quaisquer variáveis de ambiente que possam ser necessárias |
| params | Alterar os valores padrão dos argumentos da linha de comando |
| process | Ajustar configurações por processo, como containers, ambientes conda ou uso de recursos |
| profile | Criar perfis predefinidos para seu Executor |
Há muitos outros escopos que você pode estar interessado em verificar.
Você provavelmente vai querer criar um perfil personalizado. Ao fazer isso, você pode
especificá-lo em tempo de execução (-profile myProfile) e o Nextflow será executado
com base nesse perfil. Muitas vezes, seu perfil personalizado incluirá informações sobre
o executor (filas, alocações, caminhos, etc...).
Se precisar de ajuda, por favor entre em contato!
Se você estiver usando o perfil padrão (não especificou -profile 'xyz'), isso pode não ser necessário.
--cleanup_workdir
Após executar o Bactopia, você notará um diretório chamado work. Este diretório é onde
o Nextflow executa todos os processos e armazena os arquivos intermediários. Após a
conclusão bem-sucedida de um processo, os resultados apropriados são extraídos e
colocados na pasta de resultados da amostra. O diretório work pode crescer muito
rapidamente! Por favor, tenha isso em mente ao usar o Bactopia (e outros pipelines
Nextflow). Para ajudar a evitar o acúmulo do diretório work, você pode usar
--cleanup_workdir para excluir automaticamente o diretório work após uma execução
bem-sucedida.